Artificial Intelligence: The Only Intelligence Capable of Making Sense of the Massive Datasets Generated from the Tools Above (The Century of Biology: Part I.VI)
“At its most fundamental level, I think biology can be thought of as an information processing system, albeit an extraordinarily complex and dynamic one. Taking this perspective implies there may be a common underlying structure between biology and information science - an isomorphic mapping between the two - hence the name of the company. Biology is likely far too complex and messy to ever be encapsulated as a simple set of neat mathematical equations. But just as mathematics turned out to be the right description language for physics, biology may turn out to be the perfect type of regime for the application of AI.” – Demis Hassabis, CEO of DeepMind and Isomorphic Labs (1)
“Biological systems are incredibly complex and nonlinear—which makes it possible for deep learning solutions to substantially outperform models that we attempt to specify by hand with math or code.”1
Among the most miraculous aspects of our universe is the extent to which the complexity of the physics underpinning it can be distilled down to unreasonably simple mathematical equations. For instance, Einstein illuminated profound truths universal of every object from bacteria to black holes with the formula: E = mc2. Five characters were all that was needed to convey a significant chunk of how things around us work. It takes 28 characters to spell my name.
Nor was Einstein alone in uncovering this ludicrous luckiness. Here are Newton’s Laws of Motion that prescribe how force works for every single object in the universe:
1. F = 0
2. F = ma
3. F1 = -F2
Moreover, physics happens to be spoken fluently in a language (mathematics) invented (or discovered) by us humans and is therefore very well tailored to our brain’s computational quirks and capacities. These happenstances mean that a person with a high enough IQ can sit there for long enough and simply think their way to equations that describe and predict the majority of physical phenomena. A wild historical fact to bring the point home is that Einstein didn’t even cite any other people in his papers… he thought them up all by himself.
Biologists are now so well endowed. Symmetries do not abound; there are no constants. Mathematics is useless to distill biology into a set of cold, hard rules, because these are emergent, complex phenomena that interact with one another, learn from another and alter their behaviors according – not inert objects moving through space. No matter how long Einstein looked at the six billion lines of human genetic code, no matter how many strolls he took to think it through, he would never be able to decipher those As, Ts, Cs, & Gs, no less tell you what drug to make based off it or simulate biology as described in the outset.
We may have finally found the proper tools to understand the messy world of the living: massive amounts of computation applied to similarly huge datasets. This excerpt sums it up well:|In 2009, three Google scientists wrote an essay entitled The Unreasonable Effectiveness of Data. Their core argument is that in fields such as economics and natural language processing, we may be “doomed to complex theories that will never have the elegance of physics equations” but we do have a new superpower: data.
Instead of attempting to explicitly write down the rules of a program, we focus on learning the rules of the program by collecting large volumes of data that represent the solution. Creating a program to accurately classify all possible images of cats is really hard. Collecting massive sets of labeled images and learning the underlying program is actually easier.
As Karpathy writes, “It turns out that a large portion of real-world problems have the property that it is significantly easier to collect the data (or more generally, identify a desirable behavior) than to explicitly write the program.” It is fairly clear that we inhabit this portion of program space in biology.
Indeed, whereas Einstein would be powerless to make sense of biology no less simulate it as describedat the outset, doing so is explicitly the goal of CZI:
To create a virtual cell, we’re building a high-performance computing cluster with 1000+ H100 GPUs that will enable us to develop new AI models trained on various large data sets about cells and biomolecules—including those generated by our scientific institutes. Over time, we hope, this will enable scientists to simulate every cell type in both healthy and diseased states, and query those simulations to see how elusive biological phenomena likely play out—including how cells come into being, how they interact across the body, and how exactly disease-causing changes affect them.
Input into the Models: Massive Datasets
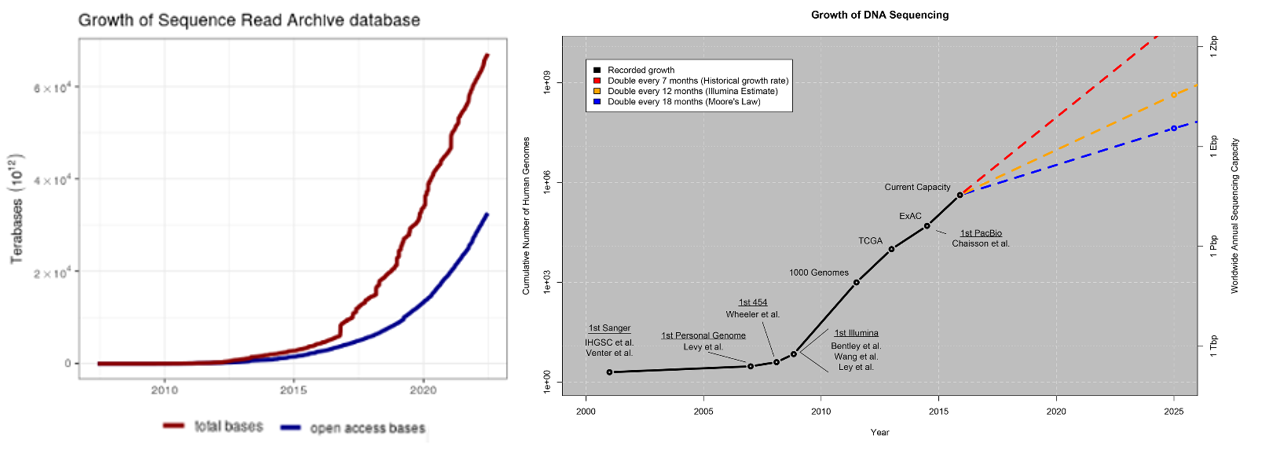
Biology, an information processing system itself, generates more data than computers could ever collect. The rate limiting factor is our ability to measure and collect data in a both accurate and high-throughput way. Every section above detailed how these capabilities are both increasing exponentially: genetics datasets, measurements of biological phenomena on scales from tissues to molecules, continuous physiological tracking with wearables on population-sized cohorts, anatomic replicas of human organs to increase the throughput of experiments. Here’s some quick stats to give a sense of how much data is out there already:
- Over the last ten years, the rate of data generated from biological experiments has 10x'd every 2 years, meaning it's 10,000x'd over the last 10 years
- A study in 2011 estimated that “the doubling time of medical knowledge in 1950 was 50 years; in 1980, 7 years; and in 2010, 3.5 years. In 2020 it is projected to be just 73 days."
- Imaging just one cell at nanometer resolution takes up the same amount of data as about 83,000 photos on your phone.
- One company, Eikon, using advanced imaging to track the behavior and movements of individual proteins in a cell puts out ~1.5PB every week. The company has 300 software engineers dedicated to applying machine learning to manage all that data.
- Another one, Recursion, has 25PB of data and has its own supercomputer to process that data. To put 25PB into perspective, every movie ever made stored in HD would be far less than that.
- The amount of genomic data available to researchers is increasing by a factor of 10 every year.
- The number of available protein sequences increased more than 20-fold in the last 5 years [2023, >2.4 billion (9, 10); 2018, ~123 million (11)].
On the last bullet, consider these points from a 2015 study:
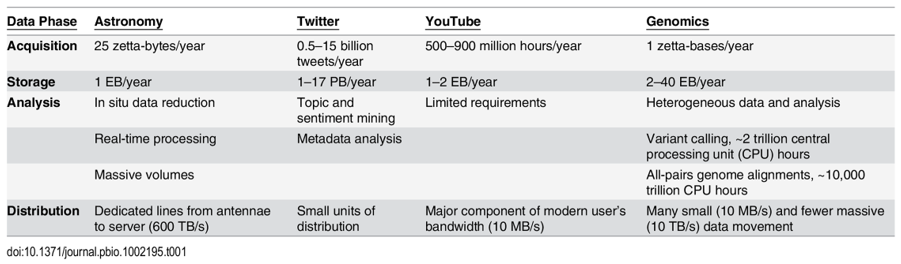
Below is a summary of a small selection of the publicly available databases with more complete catalogues of databases here, here and here.
Data availability across institutions may have just gotten a boost from ARPA-H’s newly launched project called Biomedical Data Fabric Toolbox that aims to improve the accessibility of biomedical research data by unifying research data from various health fields. The goal is to create a comprehensive search engine for various data types, enabling quicker and more precise insights into health-related questions. Initial partners include agencies like the National Cancer Institute, National Center for Advancing Translational Sciences, etc.
Now, with all this said, our data collection and aggregation can still be improved dramatically. Read this journal article for more detail on where we need to improve to make full use of AI in biology.
Summary of AI’s Progress from a High Level
As our ability to collect data has improved parabolically over the last couple decades, our ability to make sense of that data has undergone a step function in the last couple years. There are now dozens if not hundreds of publicly announced foundational models for biology. Like in areas outside of biology like language models, the list of models includes the full breadth of architectures (encoder or decoder only to encoder-decoder transformers). They’ve been applied to problems from generating proteins de novo to predicting drug development outcomes to reading MRIs. Some early applications have had tremendous success like DeepMind and David Baker’s lab effectively solving the problem of predicting protein structure from sequence or the fact there are now more than 70 AI-derived small molecules, antibodies and vaccines in clinical trials.1
Here's the link to the database of bio models, healthcare models. Consider the charts below.
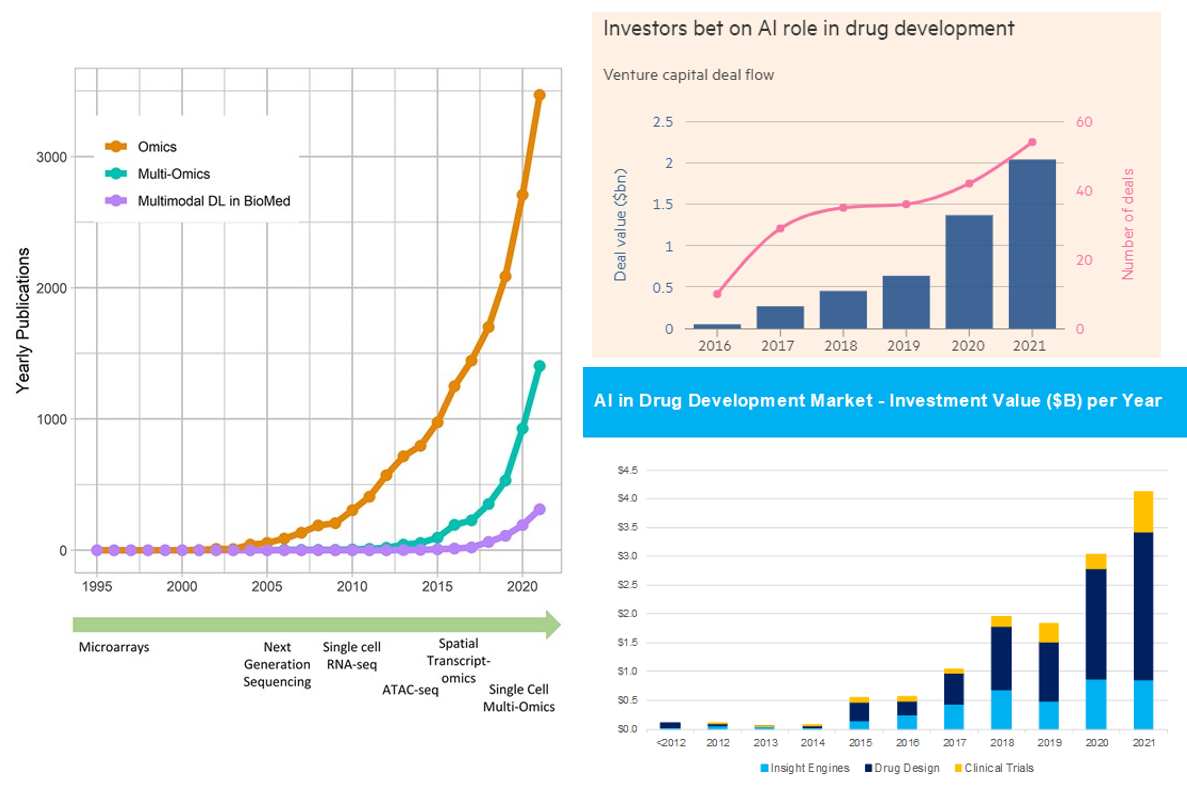
With all this said, one shouldn’t get overly carried away by the progress thus far and the prospects moving forward. Remember that computational methods have been applied to biology for decades. Indeed, here’s a good history of AI in drug discovery for that context. While the advent of foundational models in biology is truly a step function improvement on those prior capabilities, it won’t transform such a broad field with such messy data overnight.
Below, I cover six ways in which AI / ML will drive our understanding of biology forward:
- Increasing the size of the map (i.e. our understanding of foundational biology): protein-protein interactions, protein folding, etc.
- Using our foundational knowledge to guide our creation / engineering of new primitives: David Baker's lab, GenAI for protein creation, optimizing binding sites, etc.
- Scanning through collected datasets of otherwise impossibly large sizes / white spaces (10^60, New Limit, Enveda)
- Improving decision making of already understood connections: MRI imaging diagnostics, omics / multidimensional data inputs, training deep learning models on PB-size datasets,
- Throughput via automated data analysis: automated microscopy or other imaging / measurement technologies, lab automation, etc.
- Increasing knowledge abundance / access
- Increasing the Size of the Map: Expanding Foundational Knowledge about Biology (e.g. how proteins fold, how molecules interact, which genetic variants lead to disease, etc.)
AI had its moon landing moment in biology in 2020 when it effectively solved the problem of predicting protein folding based on the genetic sequence. As described in prior sections, researchers have spent half a century experimentally identifying 3D protein structures by crystalizing them and then shooting x-ray beams at them. Taking roughly five years of PhD work per protein, those one million PhD years yielded a dataset of nearly 200K solved proteins by 2020. DeepMind spent five years developing a deep learning algorithm trained on that dataset that in 2020 approached accuracy levels considered equivalent to experimental work. They used it to predict the structure of all 200M proteins – or, one billion years of PhD work, and then released the dataset to the world for free. The European Molecular Biology Laboratory’s European Bioinformatics Institute deemed 35% to be highly accurate — as good as experimentally determined structures — and another 45% accurate enough for some applications. More than 1.2 million researchers from more than 200 countries have now accessed AlphaFold to view millions of structures.
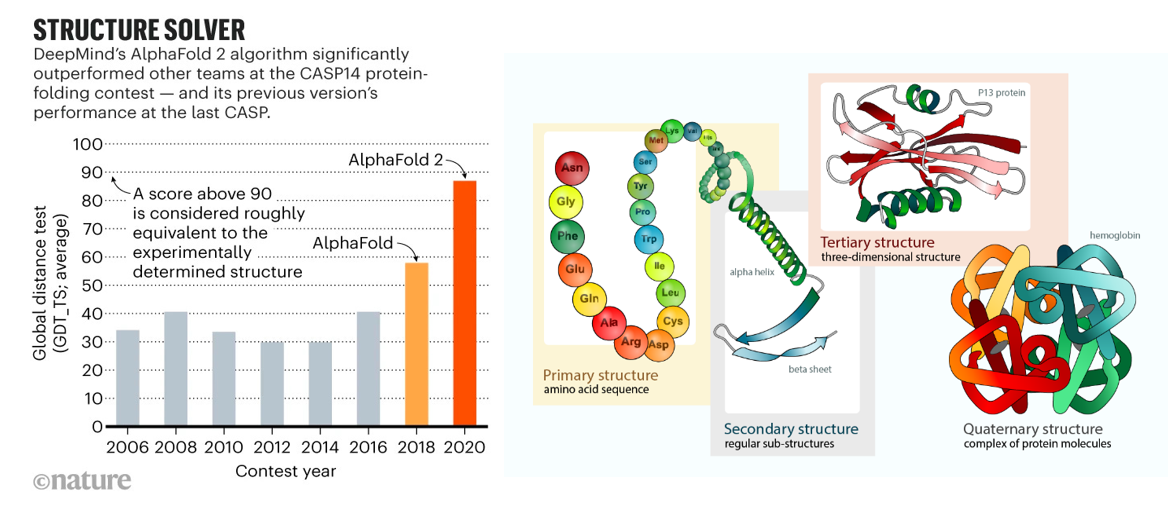
But, DeepMind is far from the only player working to predict protein structure from sequence:
- In November 2022, Meta released the ESM Metagenomic Atlas, an open atlas of ~620 million predicted metagenomic protein structures and an API that allows researchers to leverage the technology. (link)
- Researchers from Chinese universities developed a single-sequence protein structure prediction method that outperformed AlphaFold2 and RoseTTAFold by ~14% on 25 orphan proteins. However, the accuracy of the predicted models is still not high, and the researchers are exploring alternative models that might improve performance. (link)
- Predicting structure without evolutionary trees of proteins for increased performance (i.e. what seems like the protein equivalent of one-shot prediction / no context prediction):
AlphaFold21 and RoseTTAFold2 are the first two deep learning based models for highly accurate prediction of protein structures. They rely on Multiple Sequence Alignments (MSAs) as inputs to their models, which map the evolutionary relationship between corresponding residues of genetically-related sequences. They are derived from large, public, genome-wide gene sequencing databases that have grown exponentially since the emergence of next-gen sequencing in the late 2000s. It is widely accepted that MSA-dependent structural prediction tools gain 3D-positional context clues from pairs of residues that co-evolve with one-another over time, implying spatial proximity. Since MSA-dependent models are driven by evolutionary information, structure prediction applications are limited to naturally-occurring protein sequences,
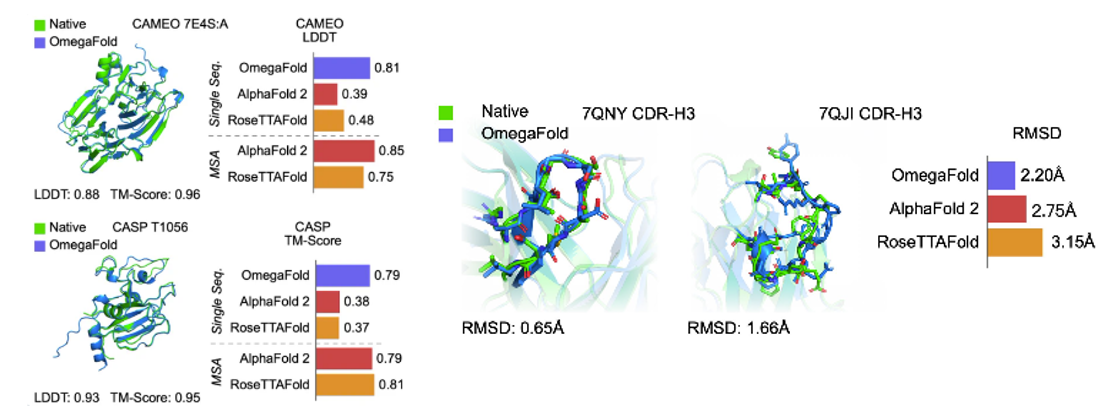
More recent tools have attempted to eliminate the need for MSAs in their predictive models by using language modeling applied on individual protein sequences (Figure 1). For example, OmegaFold3 has a language modeling component called OmegaPLM that uses transformers and attention mechanisms to learn per residue and residue-pair representations for each sequence of protein. OmegaFold3, along with other single sequence based models such as HelixFold-Single4 and Meta’s model, called ESMFold5, have higher potential in predicting structure of orphan proteins and antibody design, as they don’t require MSAs as their input. However, they have lower general accuracy for proteins with MSAs compared to AlphaFold2 and RoseTTAFold. In contrast to MSA-dependent models, the domain of applicability for language model based approaches may extend beyond naturally-occurring protein sequence for structure prediction - this may include mutated protein structure prediction or protein engineering tasks. (link)
- Without the need to search MSAs, these models are also faster. ESMFold is 60 times faster than AlphaFold2 for short protein sequences, although this difference is of lower significance for long sequences. OF takes only a few seconds for predicting a normal-size protein. (link)
Predicting folding stability for 900K proteins and testing experimentally:
- Protein sequences vary by more than ten orders of magnitude in thermodynamic folding stability2 (the ratio of unfolded to folded molecules at equilibrium). Even single point mutations that alter stability can have profound effects on health and disease3,4 , pharmaceutical development8–10 and protein evolution5–7 . Thousands of point mutants have been individually studied over decades to quantify the determinants of stability11, but these studies highlight a challenge: similar mutations can have widely varying effects in different protein contexts, and these subtleties remain difficult to predict despite substantial effort12,13
- The hidden thermodynamics of folding can drive disease3,4, shape protein evolution5–7 and guide protein engineering8–10, and new approaches are needed to reveal these thermodynamics for every sequence and structure. Here we present cDNA display proteolysis, a method for measuring thermodynamic folding stability for up to 900,000 protein domains in a one-week experiment. From 1.8 million measurements in total, we curated a set of around 776,000 high-quality folding stabilities covering all single amino acid variants and selected double mutants of 331 natural and 148 de novo designed protein domains 40–72 amino acids in length. Using this extensive dataset, we quantified (1) environmental factors influencing amino acid fitness, (2) thermodynamic couplings (including unexpected interactions) between protein sites, and (3) the global divergence between evolutionary amino acid usage and protein folding stability. We also examined how our approach could identify stability determinants in designed proteins and evaluate design methods. The cDNA display proteolysis method is fast, accurate and uniquely scalable, and promises to reveal the quantitative rules for how amino acid sequences encode folding stability (link)
Protein-protein interactions:
- Protein–protein interactions are central mediators in biological processes. Most interactions are governed by the three-dimensional arrangement and the dynamics of the interacting proteins1. Such interactions vary from being permanent to transient2,3. Some protein–protein interactions are specific for a pair of proteins, while some proteins are promiscuous and interact with many partners. This complexity of interactions is a challenge both for experimental and computational methods. Often, studies of protein–protein interactions can be divided into two categories, the identification of what proteins interact and the identification of how they interact. Although these problems are distinguished, some methods have been applied to both problems4,5. Protein docking methodologies refer to how proteins interact and can be divided into two categories considering proteins as rigid bodies; those based on an exhaustive search of the docking space6 and those based on alignments (both sequence and structure) to structural templates7. Exhaustive approaches rely on generating all possible configurations between protein structures or models of the monomers8,9 and selecting the correct docking through a scoring function, while template-based docking only needs suitable templates to identify a few likely candidates. However, flexibility has often to be considered in protein docking to account for interaction-induced structural rearrangements10,11. Therefore, flexibility limits the accuracy achievable by rigid-body docking12, and flexible docking is traditionally too slow for large-scale applications. A possible compromise is represented by semi-flexible docking approaches13 that are more computationally feasible and can consider flexibility to some degree during docking. Regardless of different strategies, docking remains a challenging problem. In the CASP13-CAPRI experiments, human group predictors achieved up to 50% success rate (SR) for top-ranked docking solutions14. Alternatively, a recent benchmark study8 reports SRs of different web-servers reaching up to 16% on the well-known Benchmark 5 dataset15 (link)
Predicting ligand-protein interactions:
- MatchMaker is a machine learning model trained to predict ligand-protein interactions, identifying potential binding between any small molecule and any protein in the human proteome. The model is particularly powerful in that it can make these predictions even with low or no data on a given protein target, because it relies on the structure of individual protein pockets, rather than that of the entire protein. As an AI-enabled platform trained on millions of known ligand-protein interactions, MatchMaker can also be used to learn biophysical patterns of proteins and ligands, adding chemical insights and improving our ability to predict a drug’s mechanism of action. (link) Here are Recursion’s studies: 1,2
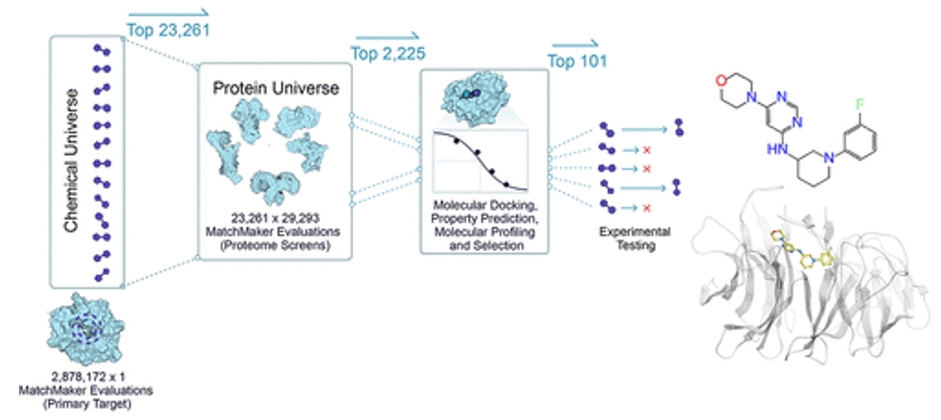
- DragonFold is another such model (link)
Limitations:
- The accuracy of ligand binding poses predicted by computational docking to AF2 models is not significantly higher than when docking to traditional homology models and is much lower than when docking to structures determined experimentally without these ligands bound. (link)
- On the surface, making the leap from AlphaFold’s and RoseTTAFold’s protein structures to the prediction of ligand binding doesn’t seem like such a big one, Karelina says. She initially thought that modelling how a small molecule ‘docks’ to a predicted protein structure (which usually involves estimating the energy released during ligand binding) would be easy. But when she set out to test it, she found that docking to AlphaFold models is much less accurate than to protein structures that are experimentally determined1. Karelina’s still not 100% sure why, but she thinks that small variations in the orientation of amino-acid side chains in the models versus the experimental structures could be behind the gap. When drugs bind, they can also slightly alter protein shapes, something that AlphaFold structures don’t reflect. (link)
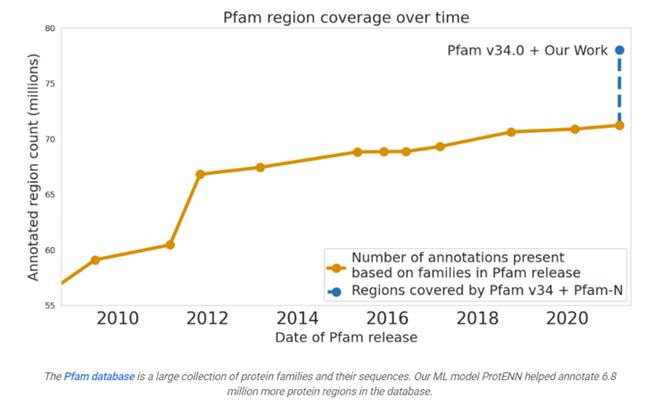
Google predicted the function of 6.8 million proteins, or the total that had been predicted over the last decade, and released the model and data to the world:
Many are familiar with recent advances in computationally predicting protein structure from amino acid sequences, as seen with DeepMind’s AlphaFold. Similarly, the scientific community has a long history of using computational tools to infer protein function directly from sequences. For example, the widely-used protein family database Pfam contains numerous highly-detailed computational annotations that describe a protein domain's function, e.g., the globin and trypsin families.
While there are a number of models currently available for protein domain classification, one drawback of the current state-of-the-art methods is that they are based on the alignment of linear sequences and don’t consider interactions between amino acids in different parts of protein sequences. But proteins don’t just stay as a line of amino acids, they fold in on themselves such that nonadjacent amino acids have strong effects on each other.
Aligning a new query sequence to one or more sequences with known function is a key step of current state-of-the-art methods. This reliance on sequences with known function makes it challenging to predict a new sequence’s function if it is highly dissimilar to any sequence with known function. Furthermore, alignment-based methods are computationally intensive, and applying them to large datasets, such as the metagenomic database MGnify, which contains >1 billion protein sequences, can be cost prohibitive.
Existing approaches have successfully predicted the function of hundreds of millions of proteins. But there are still many more with unknown functions — for example, at least one-third of microbial proteins are not reliably annotated. As the volume and diversity of protein sequences in public databases continue to increase rapidly, the challenge of accurately predicting function for highly divergent sequences becomes increasingly pressing.
Variant's impact on function based on structure:
- AlphaMissense predicted all 71 million human missense variants. Single–amino acid changes in proteins sometimes have little effect but can often lead to problems in protein folding, activity, or stability. Only ~2% of the 4 million variants have been experimentally investigated, while the vast majority of them are of unknown clinical significance. This limits the diagnosis of rare diseases, as well as the development or application of clinical treatments that target the underlying genetic cause. AlphaMissense classify 32% of all missense variants as likely pathogenic and 57% as likely benign using a cutoff yielding 90% precision on the ClinVar dataset, thereby providing a confident prediction for most human missense variants. Predictions for all single–amino acid substitutions in the human proteome are provided as a community resource.
- Century of Bio has a good summary on the work and various models leading up to this one AlphaMissense outperforms the former SoTA model published in Nature in August
How genes interact:
- Geneformer was published in Nature May 2023, pretrained from ~30 million single-cell transcriptomics, making predictions about the network of interacting genes:
A network view on genes
Many genes, when active, set off cascades of molecular activity that trigger other genes to dial their activity up or down. Some of those genes, in turn, impact other genes—or loop back and put the brakes on the first gene. So, when a scientist sketches out the connections between a few dozen related genes, the resulting network map often looks like a tangled spiderweb. If mapping out just a handful of genes in this way is messy, trying to understand connections between all 20,000 genes in the human genome is a formidable challenge. But such a massive network map would offer researchers insight into how entire networks of genes change with disease, and how to reverse those changes.
"If a drug targets a gene that is peripheral within the network, it might have a small impact on how a cell functions or only manage the symptoms of a disease," said Theodoris. "But by restoring the normal levels of genes that play a central role in the network, you can treat the underlying disease process and have a much larger impact."
They first asked Geneformer to predict which genes would have a detrimental effect on the development of cardiomyocytes, the muscle cells in the heart. Among the top genes identified by the model, many had already been associated with heart disease. "The fact that the model predicted genes that we already knew were really important for heart disease gave us additional confidence that it was able to make accurate predictions," said Theodoris.
However, other potentially important genes identified by Geneformer had not been previously associated with heart disease, such as the gene TEAD4. And when the researchers removed TEAD4 from cardiomyocytes in the lab, the cells were no longer able to beat as robustly as healthy cells. Therefore, Geneformer had used transfer learning to make a new conclusion; even though it had not been fed any information on cells lacking TEAD4, it correctly predicted the important role that TEAD4 plays in cardiomyocyte function.
Finally, the group asked Geneformer to predict which genes should be targeted to make diseased cardiomyocytes resemble healthy cells at a gene network level. When the researchers tested two of the proposed targets in cells affected by cardiomyopathy (a disease of the heart muscle), they indeed found that removing the predicted genes using CRISPR gene editing technology restored the beating ability of diseased cardiomyocytes. (link)
RNA structure prediction:
- Compared to hundreds of thousands of solved the Raphael had only 18 RNA structures to work with. Despite this serious data scarcity, Raphael developed extremely clever techniques to produce a state-of-the-art deep learning model for RNA structure prediction. In August 2021, his work, Geometric deep learning of RNA structures, landed on the cover of Science Magazine, an honor few scientists ever experience in their career. The core insight that Raphael had was that techniques from geometric deep learning would make it possible to represent the 3D coordinates of molecules in a way that was rotationally equivariant. In plain English, for any transformation to the 3D shape of the input structure, the output would be equivalently transformed. This highly creative machine learning advance made it possible to learn a state-of-the-art model with only 18 structures, a feat that most people in the field wouldn’t have considered possible. When thinking back on the initial study, Playground Global general partner Jory Bell felt that “the AI alone was impressive enough that Atomic was investable even without a data platform.” (link)
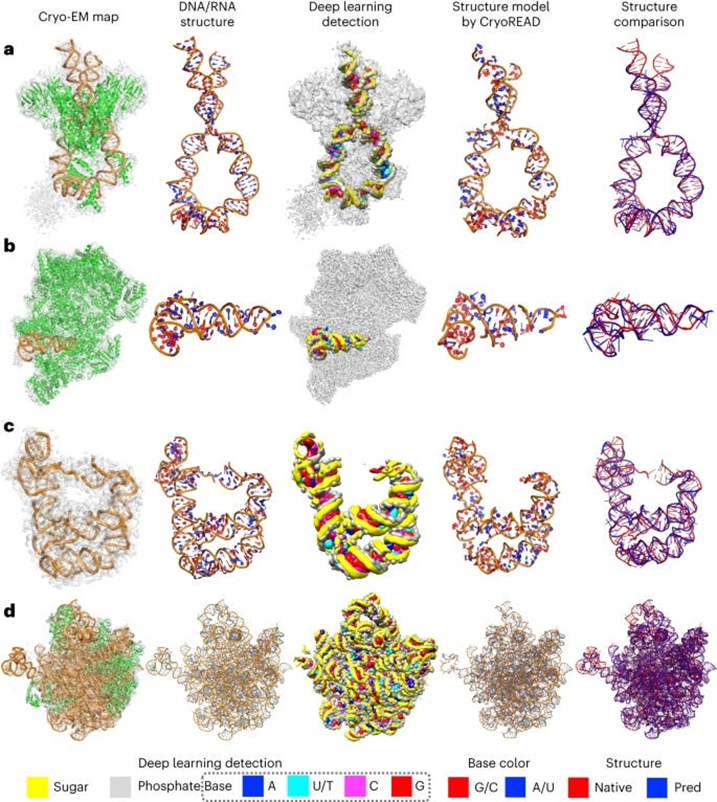
- The authors of this paper developed CryoREAD, an automated de novo nucleic acid atomic structure modeling method based on deep learning. The system works by identifying phosphate, sugar, and base positions fundamental to nucleic acid structures in a cryo-EM map with deep learning and then modeling that into a 3-D structure. The sugars are connected to form a backbone and the nucleic acid sequence is projected onto that. The system was tested at the 2-5 Å resolution, medium-scale resolution. They found that 85.7% of atoms were placed correctly within 5Å and 52.5% of nucleotides were correctly identified. The authors also tested their CryoREAD model against existing software that is mainly used for protein reads but is often adapted for understanding nucleic acids. While limitations of nucleotide sequence prediction need to be addressed, this work presents a foundation for DNA/RNA structure modeling without manual human input. This can help fill a fundamental tool gap in structural biology and modeling. (link)
RNA & protein interaction:
- In Sep ‘23, the team at Deep Genomics preprint published “BigRNA,” a foundation model for multiple predictions including protein and micro-RNA binding sites and the effect of variants. This advance was enabled by a multi-head transformer model. “Predicting protein and microRNA binding sites, tissue-specific expression, and variants impact on gene expression, splicing”
- Using our foundational knowledge to guide our creation / engineering of new primitives and therapies (e.g. David Baker's lab, GenAI for protein creation, optimizing binding sites, etc.)
Generative AI has been used for more than creating pictures of cats in space. David Baker and his lab at University of Washington called the Institute for Protein Design have spent over a decade now exploring how to use AI to design proteins and other macromolecules from scratch according to desired specifications. This, of course, would be wildly useful. Imagine drug makers knowing the exact structural details of the antigen they’re targeting and turning to the computer to create a protein de novo that binds perfectly to that receptor. These models have come a long way:
Neural networks such as RFdiffusion seem to really shine when tasked with designing proteins that can stick to another specified protein. Baker’s team has used the network to create proteins that bind strongly to proteins implicated in cancers, autoimmune diseases and other conditions. One as-yet unpublished success, he says, was to design strong binders for a hard-to-target immune-signaling molecule called the tumor necrosis factor receptor — the target for antibody drugs that generate billions of dollars in revenue each year. “It is broadening the space of proteins we can make binders to and make meaningful therapies” for, Watson says.
But early signs suggest that RFdiffusion’s creations are the real deal. In another challenge described in their study, Baker’s team tasked the tool with designing proteins containing a key stretch of p53, a signaling molecule that is overactive in many cancers (and a sought-after drug target). When the researchers made 95 of the software’s designs (by engineering bacteria to express the proteins), more than half maintained p53’s ability to bind to its natural target, MDM2. The best designs did so around 1,000 times more strongly than did natural p53. When the researchers attempted this task with hallucination, the designs — although predicted to work — did not pan out in the test tube, says Watson.
Overall, Baker says his team has found that 10–20% of RFdiffusion’s designs bind to their intended target strongly enough to be useful, compared with less than 1% for earlier, pre-AI methods. (Previous machine-learning approaches were not able to reliably design binders, Watson says). Biochemist Matthias Gloegl, a colleague at UW, says that lately he has been hitting success rates approaching 50%, which means it can take just a week or two to come up with working designs, as opposed to months. “It’s really insane,” he says.
However, they’re of course far from perfect:
Tanja Kortemme, a computational biologist at the University of California, San Francisco, is using RFdiffusion to design proteins that can be used as sensors or as switches to control cells. She says that if a protein’s active site depends on the placement of a few amino acids, the AI network does well, but it struggles to design proteins with more-complex active sites, requiring many more key amino acids to be in place — a challenge she and her colleagues are trying to tackle.
Another limitation of the latest diffusion methods is their inability to create proteins that are vastly different from natural proteins, says Yang. That is because the AI systems have been trained only on existing proteins that scientists have characterized, he says, and tend to create proteins that resemble those. Generating more-alien-looking proteins might require a better understanding of the physics that imbues proteins with their function.
That could make it easier to design proteins to carry out tasks no natural protein has ever evolved to do. “There’s still a lot of room to grow,” Yang says.
A different group of researchers released a discrete diffusion model called EvoDiff trained on protein sequence alone, as opposed to models like RFdiffusion that utilize structure data. This expands the protein design space, enabling the generation of proteins that fall outside the reach of structure-based models, including those with disordered regions.
Here’s a great summary of how they work:
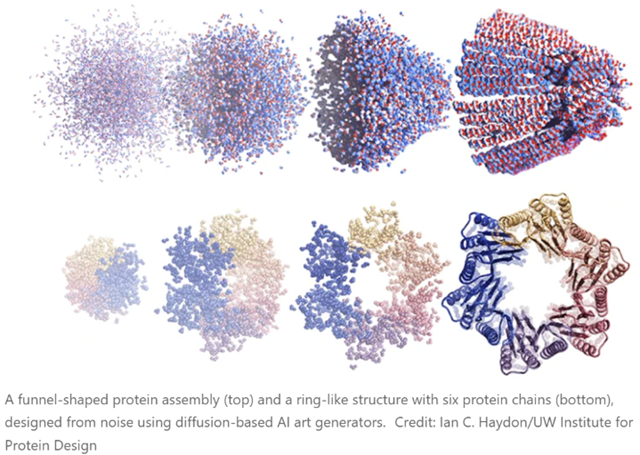
The latest methods, though, are based on the diffusion neural network approaches that are used to generate prompted images and artwork (Stable Diffusion, DALL-E, Midjourney and so on). Greatly simplified, these work by being trained on real data (photos or human-created images, in the case of the three mentioned), which then have Gaussian noise added to them (forward diffusion, and the neural network’s task is to “learn” to reverse this and produce an actual image again (denoising or reverse diffusion). This allows new images to be produced (from various types of noise!) that can resemble the images used to train the models but do not actually copy them. As many readers will be aware, this is setting off all kinds of fireworks in copyright law, as well as profoundly mixed emotions from human illustrators and artists, along reams of speculation about what we mean by words like “creativity”, “copy”, “inspiration”, “imitation”, “original”, etc.
There are no such concerns - well, not yet - with the extension of these techniques to protein design. There are several of these working right now (RFdiffusion, for example). In this case, you do the forward diffusion step with the structures of real proteins from the PDB. Then you let them grind away on de-noising until they can spit out plausible protein structures in the reverse direction. If you just hand such software a pile of noise, it generates proteins that aren’t in the PDB, but don’t seem to have anything wrong with them, and whose structures are as believable as any real one. Experimentally, about half of them turn out to be both soluble and show CD spectra consistent with the designed degree of helicity, etc. The new paper linked in the opening of this blog also shows cryo-EM structures of many proteins that match the aimed-for structures.
And if you add some spatial constraints along the way during the denoising step, you can aim the results toward specific shapes - and that includes complementary shapes to other protein surfaces. The paper tests this with the p53/MDM2 interaction, generating new replacement candidates for p53 in this pair, some of which are two to three orders of magnitude more potent binders than p53 itself. Binders to several other protein surfaces are shown as well.
Overall, Baker’s team estimates that they have about a 15% success rate with such designs, which is far, far above where things were just two or three years ago. And that rate may have already improved. The bottleneck is making and testing the proteins themselves; these techniques are spitting out so many plausible hits that it’s hard to keep up. Of course, that’s a better situation than we used to face, a long list of things where almost none of them actually worked. There are worse problems!
This ability to just come up with new proteins on demand is of course going to permanently alter protein science, to the point that it’s eventually going to be hard to explain to the young ‘uns what it used to be like without it. The possibilities for chemical biology, model systems, and eventually outright therapeutics are so numerous that it’s hard to even know where to start. But to get these to work, we’ll need to know more than a shape we’re trying to mimic: toxicity, immunogenicity, and binding to other proteins that we haven’t even studied are all factors that will have to be explored experimentally. All that knowledge (and what we know about these topics already) will surely get fed back into more advanced versions of protein-design software.
Another challenge will be attempting to design functional proteins like enzymes, or things that need to change their shape in different situations. The current diffusion models are working off more or less a static picture, and while that can take you very far indeed, it leaves a lot on the table. Extending these to more dynamic states is (as you’d imagine) a very hot topic of current research.
And keep in mind, these are all proteins that are based off of known folds and structural motifs (after all, the software was trained on the PDB). You have to wonder what else is out there: are there useful designs that evolution has never gotten around to exploring? That’s a longstanding question in the field, and answering that one is going to take abilities that we don’t have even now. But the ones we have, and the ones that we are gaining right now - they really are something to see.
The foundational AI models detailed in part 1 of this section alongside the generative AI models above have been used to better design and engineer new therapies and primitives. Here are some examples:
- The Baker Lab developed a protein engineering system that combines structure prediction networks and diffusion models. By fine tuning the RoseTTAFold protein folding algorithm on a protein structure denoising task (creating a new model called RFdiffusion), strong performance is obtained on a range of tasks including protein binder design and enzyme active site scaffolding. The experimental success rate (i.e., the proportion of experimentally synthesized and tested proteins that confirm the in silico prediction) of RFdiffusion was improved by two orders of magnitude relative to previous algorithms developed by the Baker Lab. (link) These researchers also published good work on improving binding sites.
- Dyno Therapeutics creating improved AAV vectors for drug delivery. Data released at ASGCT this year shows the difference between AAV9 and the new Dyno bCap 1 delivery vector for pan-brain transduction. Dyno claims that bCap 1 shows a “100x improvement versus AAV9 in delivery to the central nervous system (CNS) and 10x detargeting of liver after intravenous (IV) dosing, as characterized across multiple non-human primate (NHP) species.” The new vector also has a 1x increase in production efficiency. (link)
- BigHat Biosciences accelerating antibody design, Coding.bio better designing CAR-T therapies, Mana Bio doing the same for lipid nanoparticles, Atomic AI doing RNA therapeutics, Generate Biomedicine combining diffusion models with GNNs to generate full multi-protein complexes that can be conditioned to design proteins with specific properties and functions, PostEra optimizing the chemical synthesis component of lead optimization,
- Engineering an enzyme that can break down PET plastics faster than anything found in nature.
- Antibody optimization:
Getting back to that first paragraph, you can see how it would be very nice to skip all that stuff I’ve been describing, and just compute your way to a designed antibody. A vast amount of time and effort has gone into that idea over the years, and it looks like substantial progress is being made. Take this new paper, for example, which is using language models of the sort that have been very useful in protein structure and design work. The authors take some clinically relevant antibodies (which you’d think would already be pretty mature from a design standpoint) and show that they can be improved even more by these methods. Ones that hadn’t been as developed improved even more dramatically, as you’d imagine. The authors compare these to other antibody-optimization computational methods (there are quite a few!) and it looks like they outperform compared to all of them. You can get similar (or greater) improvements with rounds of biology-based optimization, but those take weeks of work in the lab as opposed to a few seconds of computation. The authors note that the evolutionary information that’s already imbedded in the structure of any sort of useful antibody seems to be what their model is working off of.
And here’s another new paper that is seeing-and-raising, in poker terms. The authors are trying for de novo antibody generation computationally, which is something that several groups have been working on. The eventual goal is for “zero-shot” examples, that is, providing an antibody for a specific antigen where there are no prior antibodies known to build off of. The team specifically removes any information about antibodies known to bind any similar proteins from the ML training set.
Specifically, they’re working on the heavy-chain complementarity-determining region 3 (HCDR3) region, which is one of these ridiculously diverse regions mentioned above (there are three of those heavy-chain regions). In this case, they took the known anti-HER2 antibody trastuzumab (sold as Herceptin) and tried to come up with completely new HCDR3 sequences based only on the sequence of the target HER2 antigen. These were substituted into the rest of the trastuzumab sequence (those other regions were all kept the same), and evaluated in a high-throughput protocol that let them test over 400,000 computed variants to see how the model did.
About 4,000 of these were estimated to show binding to HER2 at some level, and 421 of them were selected for further confirmation. 71 of these had single-digit nanomolar affinity, and three of them showed tighter binding than trastuzumab itself. This can be compared to an earlier effort from another group that was working towards something like this - in that case, though, the ML model was trained on about 10,000 sequences of trastuzumab variants, then used to screen a computational library of about ten million more. That model predicted about a million of these would bind HER2, and that team tested thirty of these at random and found that they all did have HER2 binding, so they were clearly on the right track.
But that one is more of a “supervised learning” approach, as compared to this new paper’s method, which sort of just rises up out of the bare earth of the sequence of the target. The HCDR3 sequences found in the current work are rather diverse from each other and from trastazumab itself, which is encouraging (and shows that it wasn’t just picking one motif and mining it for all it was worth). Importantly, they are also distinct from the sorts of sequences found in the training set (a key validation for such models). Modeling confirms this diversity, and also suggests that these sequences are exploiting various binding events (and not, for example, just beating hydrophobicity to death, which is also something you want to watch out for). These are all good signs!
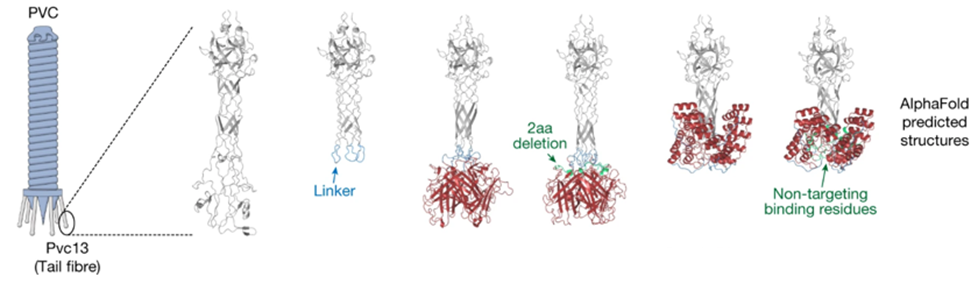
- Redesigning a drug delivery tool found in bacteria: In March this year, Feng Zhang and colleagues used AlphaFold to design a “molecular syringe” for programmable protein delivery into cells. The researchers hijacked the ‘syringe’ that some viruses and bacteria use to infect their hosts and put it to work delivering potentially therapeutic proteins into human cells grown in the laboratory. They used AlphaFold to redesign the structure such that it would recognize mouse and human cells instead of insects, as it’s originally intended for. With further optimization, the approach might also be useful for delivering the components needed for CRISPR–Cas9 genome editing. It has broad potential applications for the treatment of cancer, design and delivery of vaccines, and gene therapy. “It’s astonishing. It is a huge breakthrough,” saying Feng Jiang.
- T-cell receptor and peptide interaction prediction:
The authors have created TAPIR—T-cell receptor and peptide interaction recognizer by using convolutional neural network encoders to read in TCR and target sequences and using that to train a model to learn themes across their interactivity. TAPIR notably can train on paired and unpaired TCRs and can predict interactions against novel targets not exposed in training tasks—this breakthrough in particular has not yet been done to our knowledge. The authors demonstrated the inverse, that TAPIR can also design TCR sequences if given a target of interest. This model lays the foundation for several useful tasks in medicine including TCR prediction for diagnostics, vaccine design, specifically cancer vaccine design, and even some cell therapies. The group launched startup VCreate to commercialize these efforts last year. (link)
- Scanning through otherwise impossibly large datasets / white spaces
The process of drug discovery or identifying underlying physical processes in any biological setting is one of finding a needle in a haystack. For instance, the number of chemical compounds estimated to exist is 1060. Drug makers must identify a physical phenomenon to alter and then scan from that chemical space one that would alter the intended target in the desired way. AI has been and will only get more useful in navigating that computational task.
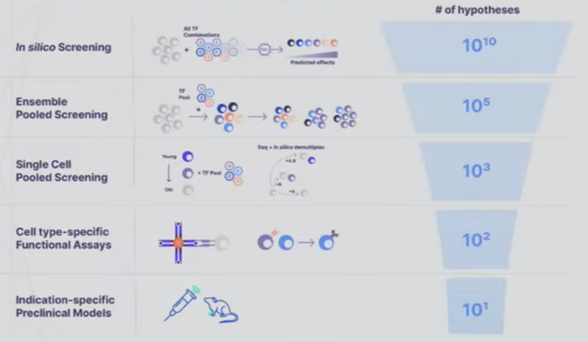
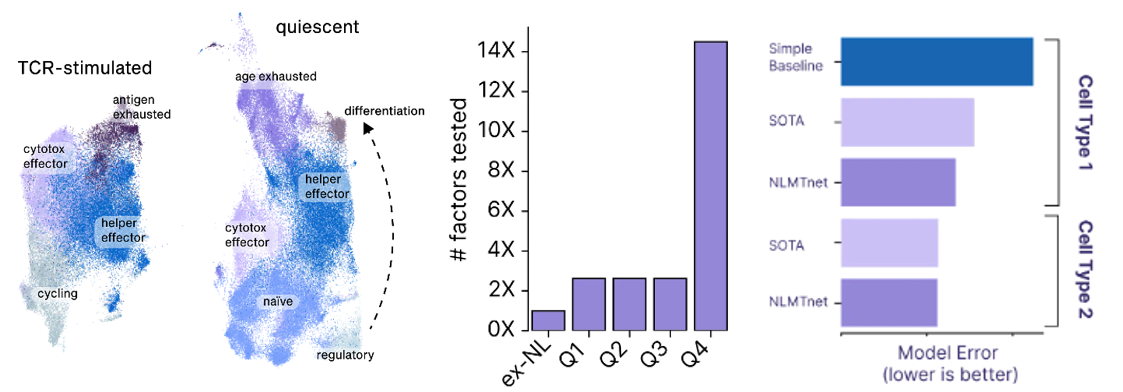
- A startup founded to cure aging called New Limit combines many of the tools detailed in the sections above to create a learning feedback loop that perfectly exemplifies the cutting edge of experiment design. They’re looking to figure out which transcription factors (TF) can epigenetically reprogram the age of a cell to a younger state without impacting anything else. Intuitively, it seems feasible to isolate age since we can already use TFs to reprogram cells to turn into different cell types (e.g. a skin cell to a lung cell) and we can reprogram both its age and its type. The tricky thing is that the combinatorial space of all possible TFs is far larger than could ever be done in the lab by perturbing a cell with a TF and seeing what happens to the cell. There are 1,700 transcription factors in the human body. Even assuming we’re smart enough to narrow that down to 400 ones that are likely to be useful for the company’s purposes, there are still 1010 combinations of 5 TFs out of 400. The traditional way of scanning a large space is to put old cells in individual test tubes, introduce the intervention, and record the results. New Limit uses advances in functional genomics to take a pool of old cells and deliver a pool of perturbations, each one getting a random combination of TFs. This allows for hundreds of thousands of combinations in a single dish where every cell represents a different experiment. But, they’re all clumped together. To figure out what actually happened, they first DNA barcode each TF perturbation. They then measure how the cell was affected not just by analyzing that one gene but by using single-cell genomics methods and ensemble chemistry to analyze all the genes in its genome, the accessibility of the genes in the genome, the proteome, etc. as well as cell type-specific phenotypes (e.g. if it’s an immune cell, does it divide and differentiation in response to a threat). They’re combining this high-throughput wet lab approach with a machine learning platform that helps them predict the effects of unseen perturbations to help determine which experiments will be most fruitful to run. These models use humanity’s understanding of TFs, the starting state of the cell, and New Limit’s prior experimental data. Very early in this feedback loop, they’ve began to map out the TF space, test experimentally 6x more TF combinations than had previously been tested, and create AI models that outperform the prior state of the art.
Model to navigate the chemical space of natural products:
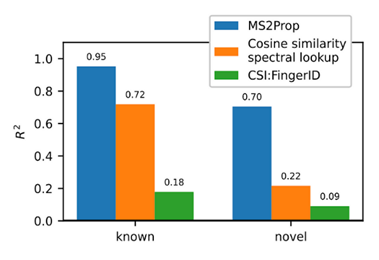
- Using transformers, we built MS2Prop: A machine learning model that directly predicts chemical properties from mass spectrometry data for novel compounds. In other words, the model predicts chemically relevant properties of compounds for drug discovery directly from mass spectra, without relying on an actual or predicted structure. As such, we can generate predictions independent of whether the compound is in an existing database. MS2Prop performance has an average R2 of 70%, meaning its predictions explain about 70% of the variation in properties from structure to structure (see preprint for list of all 10 properties) for novel compounds. This is in contrast to an R2 of 22% for the default method of looking up the closest spectral match in a database and calculating the properties from that molecule or an R2 of 9% using CSI:FingerID, a publicly accessible tool which combines fragmentation tree computation and machine learning. We show performance gains across key properties like synthetic accessibility (addressing Problem #3 above), fraction of sp3 carbons, or quantitative estimation drug-likeness (QED).
For the first time, MS2Prop enables confident decision-making about novel chemical space directly from mass spectrometry data. MS2Prop is not only significantly more accurate. It is also orders of magnitude faster (12,000 times, on average) than the state of the art. It takes just ~2 milliseconds for MS2Prop to generate a prediction from an MS2 spectrum. This performance efficiency allows us to:- Generate predictions in step with the throughput of our platform (analyze MS2 spectra related to tens of thousands of compounds daily), and
- Study unannotated natural chemical space across hundreds of millions of spectra for drug-likeness
- Improving diagnostics and clinical decision making
Healthcare as its administered currently is a high-skilled people-based service business with costs directly proportional to the number of employees. The vision here isn’t to put doctors out of work. It’s use technology to administer it over a wider swath of people. The goal is to make quality healthcare cheap and abundant in America and the world. Even in the US access to quality medical assistance varies wildly. D.C. has 3.5x as many physicians per person as Idaho. That differential between the highest and lowest density globally is 75:1 for Monaco vs many African countries. To make healthcare widespread, you can’t simply increase the number of doctors — not only is there already a shortage of doctors in the millions if not tens of millions globally but they require ~24 yrs of schooling and are among the highest paid workers so adding more is neither easy nor cheap. To scale up healthcare worldwide you need to make healthcare provision a tech-first business, weaving it into the core of the product offering.
Google’s making progress on the latter vision. A research scientist in the Google Health division gave a talk at a recent conference on their new medical-specific model Med-PaLM 2 designed to answer medical questions with maximal accuracy and safety. It’s the first LLM to perform at an “expert” test-take level on US Medical Licensing Exam questions and to pass India’s AIIMS and NEET medical exams. In the study, human physicians rated Med-PaLM’s answers better in many cases than other humans’.
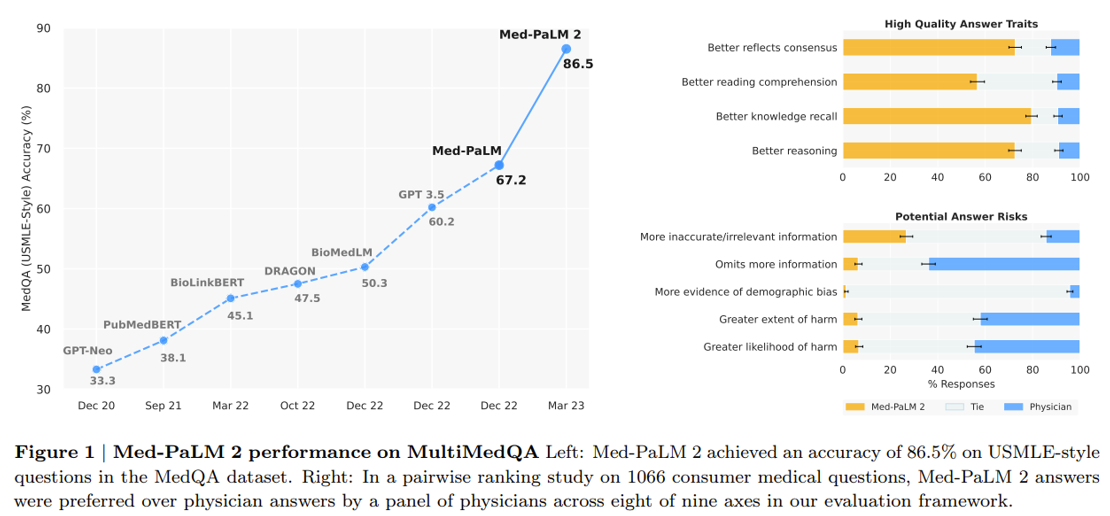
Last month, Google released Med-PaLM Multimodal a multimodal model that can reason using language, imaging, and genomics. Integrating as many types of data as possible into the training set will be vital to improving performance further.
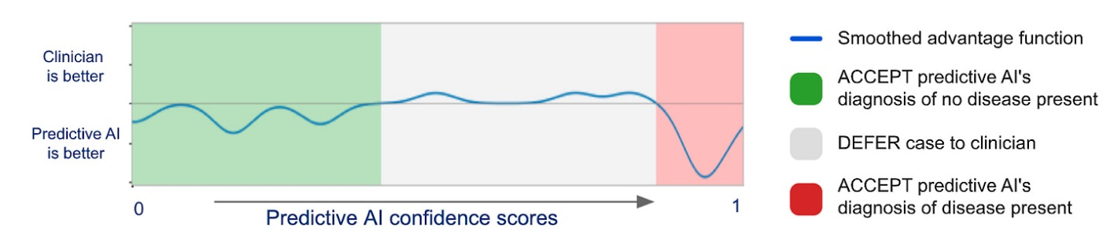
Again, this won’t replace human doctors in America. It may decrease costs of unnecessary visits and will hopefully improve doctor’s diagnosis if they chose to work with the AI. On the second point, DeepMind and Google developed a model to predict when the physician had the most accurate interpretation of medical images and when it did. They published their results in Nature showing a best-of-both-worlds:
However these technologies get integrated in the US, they’ll be amazingly beneficial in the areas of the world with internet but 1/30th of the doctors as the US. That’s the long-term vision of these teams.
- Microsoft has partnered with digital pathology provider Paige to create the world's largest image-based AI model for detecting cancer. This AI model, trained on an unprecedented amount of data, can identify both common and rare cancers, potentially aiding doctors dealing with staffing shortages and increasing caseloads. Paige has been working to digitize pathology workflows, and their FDA-approved viewing tool, FullFocus, allows pathologists to examine digital slides on screens instead of microscopes. However, digital pathology comes with significant storage costs, which Paige aims to overcome with Microsoft's help. Paige's AI model is currently the largest publicly announced computer vision model, trained on 4 million slides to identify various cancer types. (link)
- Predicting disease from retinal images: The paper introduces RETFound, a new foundation model for retinal image analysis based on self-supervised learning (SSL). RETFound was trained on a large dataset of over 1.6 million unlabeled retinal images, including both color fundus photos and optical coherence tomography scans. After pretraining with SSL, RETFound can be efficiently fine-tuned for a wide range of downstream tasks using small labeled datasets. The researchers evaluated RETFound on tasks including diagnosis of retinal diseases like diabetic retinopathy, prognosis of conditions like wet AMD, and prediction of cardiovascular diseases from retinal images. Across all tasks, RETFound achieved significantly higher performance compared to other models, even those pretrained on ImageNet. It also required less labeled data to adapt to new tasks. Qualitative analyses showed RETFound learns to identify relevant anatomical structures for each disease. (link)
- Multimodal model trained on chest radiograph images and clinical data achieved state of the art diagnostic performance
- Rapid mid-surgery classification of patient’s cancer tumor for diagnostics:
Central nervous system tumours represent one of the most lethal cancer types, particularly among children1. Primary treatment includes neurosurgical resection of the tumour, in which a delicate balance must be struck between maximizing the extent of resection and minimizing risk of neurological damage and comorbidity2,3. However, surgeons have limited knowledge of the precise tumour type prior to surgery. Current standard practice relies on preoperative imaging and intraoperative histological analysis, but these are not always conclusive and occasionally wrong. Using rapid nanopore sequencing, a sparse methylation profile can be obtained during surgery4. Here we developed Sturgeon, a patient-agnostic transfer-learned neural network, to enable molecular subclassification of central nervous system tumours based on such sparse profiles. Sturgeon delivered an accurate diagnosis within 40 minutes after starting sequencing in 45 out of 50 retrospectively sequenced samples (abstaining from diagnosis of the other 5 samples). Furthermore, we demonstrated its applicability in real time during 25 surgeries, achieving a diagnostic turnaround time of less than 90 min. Of these, 18 (72%) diagnoses were correct and 7 did not reach the required confidence threshold. We conclude that machine-learned diagnosis based on low-cost intraoperative sequencing can assist neurosurgical decision-making, potentially preventing neurological comorbidity and avoiding additional surgeries.
- Automated instruments and software to increase scientist productivity
Recall the smart microscopes mentioned in the microscopy section. Eventually, much of science will be conducted in that way: the scientist tells the instrument what data to collect and it figures out how to do it, executes it in the background as the scientist works on other things, and automatically collects and organizes the data. A clear example of how the future will look is this open-source tool using an LLM similar to ChatGPT to allow scientists to converse with an imaging instrument in natural language. Users can instruct complex tasks without any prior coding experience. For instance, a user could say “segment cell nuclei in the selected image on the napari viewer, then count the number of segmented nuclei, and finally return a table that lists the nuclei, their positions, and area.” The program can also either get or give advice on how the instructions should be carried out. A user could ask it to create a step-by-step plan for that nuclei segmentation task and the program will come back with a detailed course of action, which can be edited if needed. Other examples of tools to increase scientists’ productivity include:
- CZI developing automated annotations of microscopy data, speeding up data processing time from months or even years to just weeks they anticipate.
- Broad’s open source CellProfiler identifies objects and measures their phenotypes automatically
- Another tool employs AI-based cell-tracking pipeline for long-term live-cell microscopy, enabling the tracking of thousands of cells over many days.
- This company built one of the largest datasets of cell shapes (400M+ cells, 1B+ images), trained an AI model to categorize cells accordingly, and a physical instrument to actually sort them.
- A reinforcement learning model that predicts the growth requirements for the culturing of a new bacteria with no prior knowledge and carries out the experiments automatically. The model correctly predicts whether a media’s growth supportive or unsupportive 90% of the time and can cut experiment timeframes to two weeks, thus helping us move beyond the meager 2% of microbial species currently culturable.
- Google’s open source DeepConsensus provides on-device error correction for Pacific Biosciences long-read sequencing instruments and was used in T2T’s latest research toward comprehensive pan-genome resources that can represent the breadth of human genetic diversity.
- Automata combines cloud-based, no-code workflow scheduling software with a robotically enabled lab bench
Here’s a chart categorizing almost every software offering in biology, many of which have an automation component.
Note that it will be a while until robots can run entire biological experiments without significant oversight from people due to their complexity and subtlety. An illustrative example of this is it took one researcher six months to realize his lab’s project was failing because a chemical leaching out of a rubber gasket in one of their cell culture wells was interfering with the enzymatic reaction. Until the robots can not just pick up the petri dish but also notice that an oily residue wasn’t supposed to be there and problem solve next steps, end-to-end automation will be elusive.
With that said, ultimately, that day will come, and experiments will be run fully autonomously. Consider the mission statement of this brand-new company founded by a very legit scientist / entrepreneur with backing from Eric Schmidt:
Today, we are announcing Future House, a new philanthropically-funded moonshot focused on building an AI Scientist. Our 10-year mission is to build semi-autonomous AIs for scientific research, to accelerate the pace of discovery and to provide world-wide access to cutting-edge scientific, medical, and engineering expertise. We have chosen to focus on biology because we believe biology is the science most likely to advance humanity in the coming decades, through its impact on medicine, food security, and climate.
Biology research today is set to scale. New techniques allow us to test tens or hundreds of thousands of hypotheses in a single experiment. New tools allow us to design thousands of proteins computationally in parallel. However, the fundamental bottleneck in biology today is not just data or computational power, but human effort, too: no individual scientist has time to design tens of thousands of individual hypotheses, or to read the thousands of biology papers that are published each day.
At Future House, we aim to remove this effort bottleneck by building AI systems – AI Scientists – that can reason scientifically on their own. Our AI Scientists will augment human intelligence. In 10 years, we believe that the AI Scientists will allow science to scale both vertically, allowing every human scientist to perform 10x or 100x more experiments and analyses than they can today; and laterally, by democratizing access to science and to all disciplines that are derived from it.
Or, take this Nature paper abstract from a collaboration between the Lawrence Berkeley National Lab and DeepMind that created an entirely autonomous material science laboratory:
To close the gap between the rates of computational screening and experimental realization of novel materials1,2, we introduce the A-Lab, an autonomous laboratory for the solid-state synthesis of inorganic powders. This platform uses computations, historical data from the literature, machine learning (ML) and active learning to plan and interpret the outcomes of experiments performed using robotics. Over 17 days of continuous operation, the A-Lab realized 41 novel compounds from a set of 58 targets including a variety of oxides and phosphates that were identified using large-scale ab initio phase-stability data from the Materials Project and Google DeepMind. Synthesis recipes were proposed by natural-language models trained on the literature and optimized using an active-learning approach grounded in thermodynamics. Analysis of the failed syntheses provides direct and actionable suggestions to improve current techniques for materials screening and synthesis design. The high success rate demonstrates the effectiveness of artificial-intelligence-driven platforms for autonomous materials discovery and motivates further integration of computations, historical knowledge and robotics.
- Increasing knowledge abundance and access
There is a phenomenon that all biologists will be aware of, where after working on a new idea for 2 years, you one day come across a paper from 2008 and say, “oh my god, if only I had known this two years ago.” If we want biology to move fast, we need to figure out how to eliminate this phenomenon.
Reading every newly published paper even in one’s narrowly defined field is impossible due to the sheer vastness of the biomedical literature. A study in 2011 estimated that “the doubling time of medical knowledge in 1950 was 50 years; in 1980, 7 years; and in 2010, 3.5 years. In 2020 it is projected to be just 73 days."
How much progress we can make in biology in the next hundred years will depend on the extent to which the language models are able to solve these problems. The primary questions here will be: what fraction of knowledge in the world can be generalized from knowledge already in the literature, and how valuable is literature synthesis when a large portion of it is incorrect? The problem of summarization will be solved by language models soon. Within a few years at most (and maybe in a few months), every lab will have immediate access to the world’s expert in all of biology, which will happily educate them about the state of the art and the relevant subtleties in whatever field they choose. Simply tell the language model exactly what you want to do, and it will summarize for you everything relevant that is known, thereby avoiding the “if only I had known this” problem. It seems to me that the unreliability of the literature, however, will mean that for the foreseeable future (essentially until we have fully parameterized lab automation), AIs will be better at suggesting experiments and interpreting results than they will be at drawing conclusions from existing literature.
Some tools already exist to increase knowledge abundance and access:
- Connected Papers is a tool to visualize all the papers ever published that are related to the one you’re currently reading.
- Several groups have been working on knowledge graphs to allow researchers to query that knowledge held in that group or the literature at large. For instance, Stephen Quake’s group embedded all 155K papers citing its work and 10K automatically generated transcripts of their internal meetings and big pharma companies have internal knowledge graphs for their proprietary datasets
- Elicit facilitates the efficient exploration of free access scientific literature and Paper Explainer summarizes a given paper and allows the user to ask questions about it
- Meta released a new model that can store, combine and reason about scientific knowledge
- CZI’s cell database searchable with natural language
Read next section: introduction to Part II