The Failures and Futures of Cancer Vaccines
The cancer vaccine field has yet to become a systematic science despite decades of work and a steadily rising number of clinical trials since the first clinical PoC a decade ago.
A core problem facing cancer vaccine development lies in the nature of neoantigens themselves. In infectious diseases, the antigenic target is unambiguously foreign to the host. With cancers, the neoantigens arising from somatic mutations are often overwhelmingly similar to normal cellular proteins. A single amino acid substitution may distinguish between "self" and "almost-self."
Intriguingly, the field has some tentative early successes in pancreatic cancer, kidney cancer and a range of solid tumors. Moderna’s upcoming Phase III readout may make it another hot me-too space.
In its Phase II readout, Moderna’s mRNA-4157 therapy in combination with Keytruda significantly improved outcomes for patients with resected stage III/IV melanoma. 18 month RFS improved to 79% vs 62% for monotherapy. For its promise, the therapy got Breakthrough Therapy Designation and $450M in upfront cash from Merck starting in 2016. That team at Moderna is perhaps the only on earth that knows how to design a working cancer vaccine. Unfortunately, they won’t respond to your emails no matter how persistent you are.
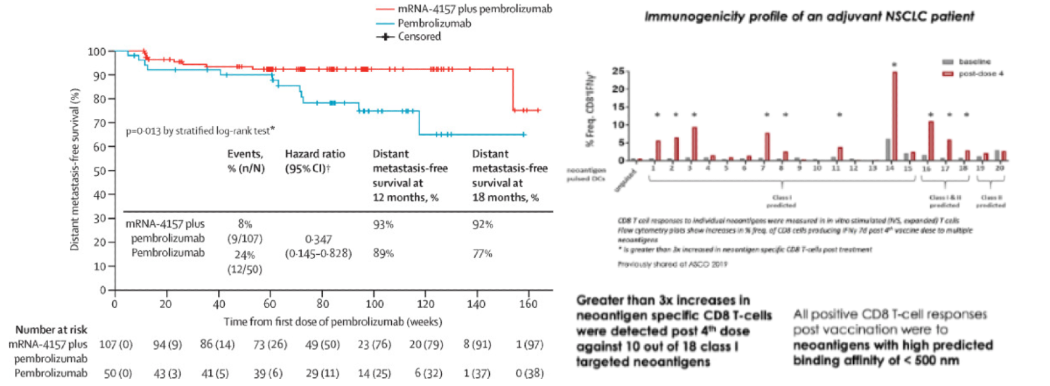
However, even that study has major caveats. The control arm lacked an adjuvant (which alone could possibly explain all of the clinical benefit), the p-value was 0.053, the RFS effect size was small, the vaccine was given to those with minimal disease burden, and melanoma is among the most immunogenic cancers.
Until results from the Moderna Phase III trial come out in 2028 and the world starts to get let into their secrets, it’s a good time to speculate on why the broader field has failed and the next-gen approaches we at Compound are excited about.
The Field’s Stumbling Blocks
Modality and Parameter Benchmarking
Despite decades of research, we don’t even know what modality to use or broad design parameters to deploy because no systematic benchmarking exists comparing mRNA, peptide, DNA, and dendritic cell vaccines. Industry lacks incentives to pay for head-to-head comparisons, while academic groups lack the resources.
The closest thing I’ve seen to systematic in-vivo modality benchmarking was done within mice and has so far tested formulations of peptides, adjuvants and mRNA with intentions to test DC and DNA vaccines in the future. The test did find that mRNA and peptide + adjuvant produce the strongest immune responses. And I've heard from multiple experts that have led major cancer vaccine programs that they internally benchmarked different modalities and found that mRNA works best.
Furthermore, basic parameters remain unstudied like optimal number of antigens, the importance of targeting multiple HLA alleles, and the balance between breadth and immunogenicity.
Prediction Algorithms
We’re still stunningly bad at predicting what neoantigens will prompt a therapeutically relevant immunogenic response.
The most comprehensive and unbiased benchmarking of neoantigen prediction capabilities came from a 2020 TESLA paper in which a consortium of 25 teams submitted predictions on a unified dataset. They tested the 608 highest ranked epitopes amongst 100K+ total predictions. Just 6% of those highest confidence predictions (0.04% overall) turned out to be immunogenic.
Even BioNTech struggles to design an effective therapy, despite its expertise in mRNA therapies, its proprietary dataset of 18,000 neoantigens collected across 4 in-human trials in addition to all its pre-clinical work, and selection criteria that sound reasonable. For their most successful trial, just 11% of the vaccinated neoantigens clearly evoked a TCR and in 4 of 16 individuals just one neoantigen drove an immune response.
It’s unclear if this low hit rate is acceptable. While the trial showed improving patient outcomes, it sorely lacked reasonable controls. Regardless, as long as hit rates are low, the effectiveness of an exorbitantly expensive therapy will vary wildly and unpredictably depending on the patient and tumor. It may work if the neoantigen that happens to work is a driver gene or if it or the HLA that expresses it can easily be adaptively lost. But it’ll often fail to contain evolutionary escape. Moreover, recent work on immunodominance suggests that vaccinating against something that is potentially immunogenic but not active against the target could be harmful.
Basic Biology and Prediction of Presentation
For a vaccine to matter, a neo-peptide must follow a complex chain of events with many failure modes that aren’t well understood. This process includes variant expression, peptide processing, transport and presentation of HLAs on the cell surface at a high enough level for T-cell receptors (TCR) to recognize. It then needs to exist in the background of stochastic TCR generation in the thymus, such that T-cells that are generated by humans would actually bear TCRs that would recognize it.
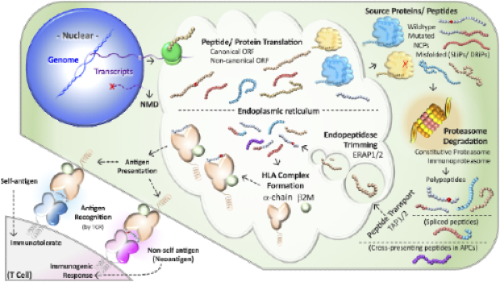
These are still incompletely understood processes from a basic science perspective and certainly can't be readily predicted a priori by algorithms.
Biological Validation
A systematic failure across the field is the reliance on surrogate immunological endpoints without validating actual tumor antigen presentation. Demonstrating T-cell responses via peptide-MHC tetramers, ELISpot assays, or other ex vivo methods does not guarantee clinical relevance. If the tumor does not actively present the targeted antigen, vaccine-induced T-cells are functionally equivalent to flu-specific T-cells in a cancer patient: present but clinically irrelevant.
Tumor Heterogeneity
Most neoantigen prediction pipelines are based on bulk sequencing data and its tissue-level averages. A vaccine targeting antigens present in only the dominant clone may successfully eliminate the bulk of the tumor while leaving resistant subclones intact, ultimately leading to treatment failure through tumor evolution.
Technologies to Turn the Tide
One expert practitioner described cancer vaccines as “a basic science project that a lot of people think is already solved.” Excitingly, a handful of technologies could quite possibly bridge that gap within a startup’s pre-clinical budget of 3-4 years and ~$10M.
While most pipelines include just short-read exome sequencing, single-cell long-read RNA sequencing could help find all the antigens’ genomic sources in their full heterogeneity. It could even discover novel targets by detecting fusion, mis-splicing, and subclonal SNV/indel sources missed by short-reads. CureVac is a rare example that does whole genome, long-read sequencing and found a novel class of neoantigens.
If desired, one could add on rich multi-omic layers like whole-exome/genome sequencing, RNA-seq, Ribo-seq and proteogenomics to expand and customize the search TSA space to include canonical coding regions, non-canonical ORFs, transposable elements, defective ribosomal products and spliced peptides.
Low-input, high-depth LC–MS/MS workflows like NeoDisc coupled with improved antibody or recombinant HLA enrichment can now retrieve 50,000-plus unique peptides from 5 × 10⁷ cells, enabling scaled surface presentation validation. Surprisingly, mass spec immunopeptidomics still remains nonconsensus within the field.
To then confirm that the neoantigens are actually triggering a therapeutically relevant tumoral response, the team should employ parallel functional testing. For instance, DNA-barcoded minigene or IVT mRNA libraries combined with 10x scTCR-seq to screen 1,000+ candidate epitopes per patient for actual T-cell activation within a week, collapsing the discovery cycle.
Such a pipeline as described above can then be fed into state of the art algorithms. This can include high dimensional deep learning models but should incorporate the lessons that the field has gleaned from antigen prediction so far. For instance, the TESLA benchmarking study found the following filters to be most effective:
- First filter for strong presentation features: MHC binding affinity < 34 nM, tumor abundance > 33 transcripts per million, binding stability > 1.4 hours, avoid mutations at position 2 of the peptide
- Second, incorporate recognition features: low agretopicity (< 0.1), high foreignness
Note that these suggestions are preliminary and mostly illustrative, with still much uncertainty remaining. A key milestone will be a pipeline that can predict neoantigens that bind not only to MHC class proteins I to activate CD8 cells which are now reasonably well understood, but also the trickier MHC class II molecules to simultaneously stimulate CD4 helper T-cells.
A strong argument is to be made that this SotA pipeline should be pointed at discovering neoantigens in addition to or other than the traditional SNVs as these are relatively low-value self-like targets. As a top lab said, “the targets are nearly self, the ML misses antigen processing so <10% of predicted pMHCs are really on the tumor, probably most actually present pMHCs are subclonal - providing easy escape, and immunogenicity is 100x+ too low.”
Uncharted science awaits as current FFPE short-read exome sequencing assays only profile ~1% of the genome.
The startup’s high-throughput pipeline could be tasked with rapidly trying to identify structural variants, frame-shift mutations, fusions, taking into account adjacent normal tissue's effect on antigen presentation, repeat elements from the dark genome, novel open reading frames, etc.
While they occur less frequently, these non-missense mutations can be disproportionately more immunogenic and are far easier for the immune system to differentiate as non-self.
Making a pipeline for non-traditional variant calling work may even require building a new bioinformatics stack.
Crucially, the team must innovate on all these dimensions at once in a unified trial because otherwise each patient may experience a different dominant failure mode. With some patients, you won't find the antigens of the right genomic source, others you won't accurately predict which are presented, while still others won’t have the right TCR repertoire in the circulating T-cell in order to engage those antigens. With more incrementalism, the field will continue along with serial trial-and-error experiments with limited conclusions.
If a team solves all these issues plaguing the field, one unavoidable issue remains. The manufacturing process as seen below is so complex for personalized cancer vaccines that they will likely cost $150K+ per patient. And that’s with Moderna having a reasonably sophisticated manufacturing process whereby bays of single-use personalized RNA machines, each the size of a refrigerator, crank out personalized mRNA sequences. A mixing device then encapsulates the mRNA in fatty nanoparticles to enhance its stability and cellular uptake.
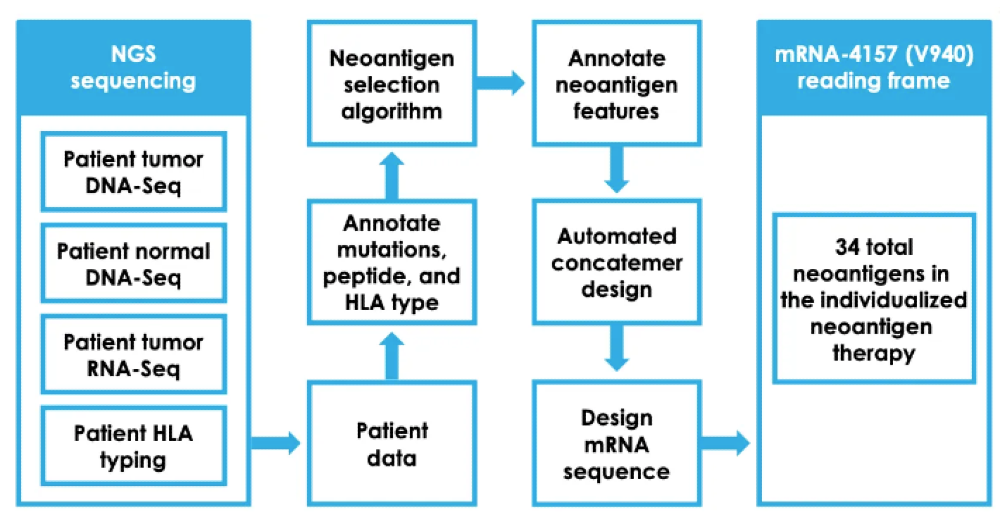
Such an expensive second-line combination vaccine may well have a hard time earning scaled usage and payment, even with Keytruda rolling off patent in 2028 and its price dropping by 30-60% from $150K/year over time.
We at Compound dream of technologies like microfluidics or cell-free synthesis replacing Moderna’s expensive machinery to manufacture personalized vaccines far more cheaply and in a decentralized manner at or near the hospital.
In addition to the above ways to make cancer vaccines in particular more effective, there’s of course many branches of work towards improving mRNA therapeutics more broadly including ways to make it more stable (e.g. circular mRNA, self-amplifying mRNA, multi-tailed mRNA), more immunogenic adjuvants, novel mRNA chemistries, etc.
All together, the vaccine must maximize three variables simultaneously: presentation probability, TCR availability, and clonal breadth across all metastatic sites. For most rigorous assessments of actually differentiated efficacy over controls, preclinical teams should strive to meet this professor’s checklist:
- Treatment of properly developed, already injected tumors
- Testing on multiple tumor models with consistent results
- The vaccine uses endogenous, tumor-specific antigens not xeno-antigens
- Control for the adjuvant
- Benchmarking relative to various formulations and modalities
What’s On the Other Side of the Rainbow
Cancer vaccines may prove to be a potent combination therapy with checkpoint inhibitors. Keytruda depends on harnessing whatever T-cells already exist in the body’s depleted pathogenic state, whereas cancer vaccines train new T-cells to spring into action and proactively protect against the tumor. Intuitively, they could possibly become standard of care together.
First they’ll largely be deployed post-resection whereby the removed tumor sample is assayed and used to design a personalized vaccine to train the body to proactively protect against recurrence. For this use case, time is of the essence and turn around times must be measured in weeks.
Who knows maybe we could even develop algorithms that predict tumor evolutionary escape and select the optimal mix of neoantigens to protect against it.
Over time, though, we will hopefully push towards the most exciting future of developing broadbased cancer vaccines by using neoantigens common across many tumors. They could come to market ~10x cheaper than personalized approaches, making the therapy possibly useful for all cancer patients and removing the payor risk.
They could even fundamentally transform how we think about cancer treatment by introducing the concept of preventative cancer vaccination.
At risk groups like those with p53 or BRCA could be vaccinated well before that demographic typically experiences disease onset. While this paradigm is easily a decade away since the vaccines not only need to be proven efficacious but also unusually safe to be used in preventative settings, this could massively expand the addressable audience as well as turn it into a regularly dosed drug.
It's possible this paradigm might start in a disease like myeloma where patients are known to have pre-cancer, are proactively identified and continuously monitored. Would-be clinical trial operators know where such patients are and their historical progression rate (~50% progress to myeloma within five years). That could possibly yield a feasible trial.
Broad-based approaches typically encode classic driver genes like KRAS, MICA/B or G12D but some groups are trying to discover novel targets for example by looking at cancer-specific splicing events (neojunctions) expressed ubiquitously across several cancer types.
With that said, it has yet to be proven possible, and cancer has only a few known exceptions that prove the rule of heterogeneity. If the difficulty of universal COVID vaccines with the 200+ unapproved clinical candidates is any guide, the goal may be frustratingly elusive.
At this point, there’s only questions and few answers. How broad the vaccine could be: major cancer types like breast cancer, subtypes like HER2 breast cancer, or high dimensional groups of tumors that would still need to be deeply sequenced? What are the minimum number of shots patients would need and how long could immunity last?
It also may be the case that a combination of targeting driver genes and more subtype or personalized neoantigens is maximally effective.
Ultimately, by pushing the frontiers of each technology discussed in the prior section as well as their integration, the startup could earn an initial wedge amongst the existing giants of Moderna and BioNTech that are commercializing 10-year-old technology.
Beyond building a more robust neoantigen prediction pipeline, the startup could outcompete incumbents by exploring novel target areas outside of the field’s almost singular focus on SNVs. From the first successful candidate, the data and know-how may readily transfer to the next.
While this field may be slowed in the short-term by regulatory changes, we at Compound are thrilled by the prospect that a startup could build a platform that sequentially addresses these ever-larger markets and clinical impact, potentially even flipping the framework of cancer care towards one of prevention from prayers for late stage metastases.