Visualizing Biology with Microscopy (The Century of Biology: Part I.III)
Since their invention in the late 16th Century, microscopes have been one of the quintessential instruments of the biological sciences. When you imagine a white lab coat biologist, chances are you think of them stooped over a microscope looking at a sample of cells. In fact, the invention of the microscope led to the discovery of the existence of cells several decades later. In the centuries since then, they’ve gotten a whole lot better.
In particular, within the last two decades, we’ve developed tools to see the entire spectra of life from individual atoms to mapping entire organs. Some of those use light waves to peer within side cells, something previously thought impossible based on our understanding of physics, while other approaches use particles not named photons, like electrons or x-ray beams. Moreover, improvements in preparation techniques like fluorescence microscopy in which glowing beads are attached to individual proteins has allowed us to light up the otherwise dimly lit theatre of life. For instance, the image on the below uses fluorescence paired with super resolution light microscopy (invented in 2009).
We’ve used these advances to map out cells, bridge the gap between morphological observation and functional profiling, track them over time, study them alive, study the interactions and mechanics of macromolecules, identify the structural configurations of those macromolecules at atomic precision.
Over the course of this section, we’ll zoom out millions of times from the narrow apertures of individual atoms to tissue level formations.
- Molecular Frame (0.1nm to 10nm)
All imaging technologies, whether they be your eyes or electron-based microscopes, work by shooting particles at an object (light photons for our eyes) and observing how they bounce off it. Intuitively, there’s a fundamental relationship between the width of the particle’s wavelength and its ability to measure that thing. Imagine a boat doing zigzags down a river; the odds that it hits a randomly placed buoy is higher the narrower those zigzags are. The narrowest wavelength of light, ultraviolet, is 400nm. In 1873, Ernst Abbe figured out that a microscope’s resolution is limited to half the wavelength of the particle it’s working with, expressed through the wonderfully simple equation called Abbe’s Limit: dmin = λ/2. That means that light couldn’t be used for cellular biology because its half-width is the size of the whole cell – the thing it’s trying to peer within.
However, the simplicity of Abbe’s equation rested on several assumptions: “a single objective lens, single-photon absorption and emission in a time-independent linear process at the same frequencies, and uniform illumination across the specimen with a wavelength in the visible range. So often Abbe's limit has been regarded as inviolable, largely because the equation is simple to understand and is powerful through being cited so often without question that it is regarded as absolute.” (link)
Abbe’s limit held steadfast for 136 years until 2009, when Stefan Hell forced this branch of physics to revisit its assumptions. He built a light-based microscope that broke through the 200-nm barrier that year. Three years later, he surpassed 70nm with new technologies called STED and PALM/STORM, allowing enabling the visualization of individual neurons in a mouse brain. Now, his latest technique, MINIFLUX, is down to singular nanometers. “We have routinely achieved resolutions of a nanometer, which is the diameter of individual molecules – the ultimate limit of what is possible in fluorescence microscopy. I am convinced that MINFLUX microscopes have the potential to become one of the most fundamental tools of cell biology. With this concept it will be possible to map cells in molecular detail and to observe the rapid processes in their interior in real time. This could revolutionize our knowledge of the molecular processes occurring in living cells.”
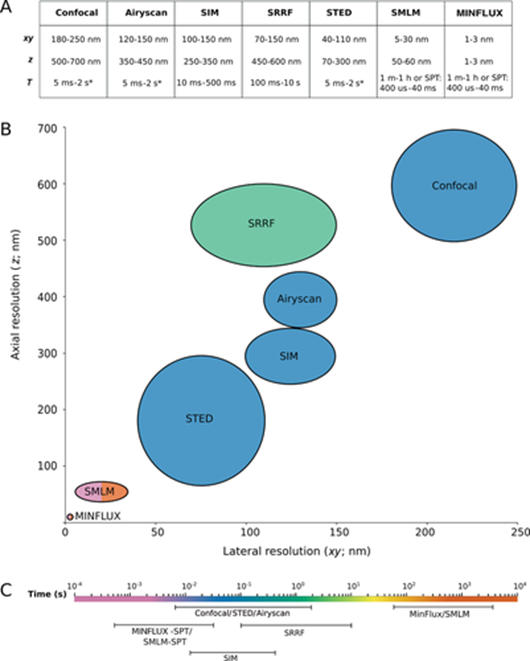
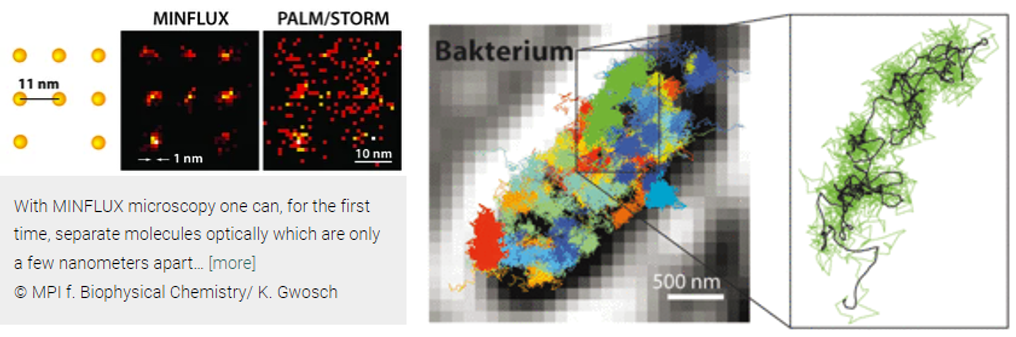
Here's how the techniques work (feel free to skip if uninterested):
Both STED and PALM/STORM separate neighboring fluorescing molecules by switching them on and off one after the other so that they emit fluorescence sequentially. However, the methods differ in one essential point: STED microscopy uses a doughnut-shaped laser beam to turn off molecular fluorescence at a fixed location in the sample, i.e. everywhere in the focal region except at the doughnut center. The advantage is that the doughnut beam defines exactly at which point in space the corresponding glowing molecule is located. The disadvantage is that in practice the laser beam is not strong enough to confine the emission to a single molecule at the doughnut center. In the case of PALM/STORM, on the other hand, the switching on and off is at random locations and at the single-molecule level. The advantage here is that one is already working at the single-molecule level, but a downside is that one does not know the exact molecule positions in space. The positions have to be found out by collecting as many fluorescence photons as possible on a camera; more than 50,000 detected photons are needed to attain a resolution of less than 10 nanometers. In practice, one therefore cannot routinely achieve molecular (one nanometer) resolution.
Hell had the idea to uniquely combine the strengths of both methods in a new concept. “This task was anything but trivial. But my co-workers Francisco Balzarotti, Yvan Eilers, and Klaus Gwosch have done a wonderful job in implementing this idea experimentally with me.” Their new technique, called MINFLUX (MINimal emission FLUXes), is now introduced by Hell together with the three junior scientists as first authors in Science.
MINFLUX, like PALM/STORM, switches individual molecules randomly on and off. However, at the same time, their exact positions are determined with a doughnut-shaped laser beam as in STED. In contrast to STED, the doughnut beam here excites the fluorescence. If the molecule is on the ring, it will glow; if it is exactly at the dark center, it will not glow but one has found its exact position. Balzarotti developed a clever algorithm so that this position could be located very fast and with high precision,“making it possible to exploit the potential of the doughnut excitation beam.”
MINIFLUX improved on STED’s ability to record real-time video recording of the inside of living cells by increasing its resolution by 20 times and its ability to track the movement of molecules by 100 times. Hell used his new toy to visualize in 3D the movement of ATP in real-time, revealing new detail about its motion. He subsequently showed it could even track such small particles amidst the normal hustle and bustle of living cells. It’s like watching a movie of our cells at their most infinitesimally small scale.
In the new study1, published in March, Hell’s group tested a version of MINFLUX that pulses linear lasers in two directions in the focal plane in quick succession, localizing the protein by finding where the overlapping fluorescence intensities are lowest. By combining multiple measurements, the researchers were able to produce tracks that show where the molecule is moving along the microtubule, like an app that maps out a runner’s path.
Although the new MINFLUX wasn’t much more effective than the previous iteration, Hell says, his team was able to use it to deduce a walking speed for kinesin of 550 nanometres per second and a stride length of 16 nanometres at ATP concentrations similar to those found in living cells. By tagging and tracking different parts of the protein, the team also showed that each stride comprises two 8-nanometre substeps, and that a kinesin’s stalk rotates as it moves, resulting in a forward motion that twirls slightly to the right. The authors also found that ATP is taken up when only one foot is bound to the microtubule, but is consumed when both feet are bound — resolving previously contradictory findings7,8. (link)
Despite achieving sub-nanometer spatial resolution, MINIFLUX “is not yet at the limit of its performance.” Improvements will come to its temporal performance with better tracking systems. Chemists aim to attach fluorescent small molecules, ideally just a couple atoms thick. New techniques in development like genetic code expansion, where synthetic amino acids are embedded into proteins, will help achieve the vision, but they’re not quite ready for the big screen.
Such advances could open multiple research avenues, especially if they allow researchers to track several proteins, or several sites in proteins, at once. This year, researchers introduced a tool called RESI (resolution enhancement by sequential imaging), which aims to do just that9. RESI can label adjacent copies of the same target molecule with different tags, allowing scientists to distinguish between molecules that are less than one nanometre apart. Although RESI currently works only on fixed or stationary molecules, combining its findings with MINFLUX tracking data on non-fixed copies could yield complementary findings about a protein’s arrangement and motion. Ries is interested in studying other motor proteins, and in a supplementary experiment shared in his paper2, applied MINFLUX to the muscle-contracting protein, myosin. In 2019, Lavis co-founded a company to use single molecule tracking for drug development. Other scientists have suggested everything from transcription factors and DNA-binding proteins to cohesin and other molecular motors as targets for future study.]
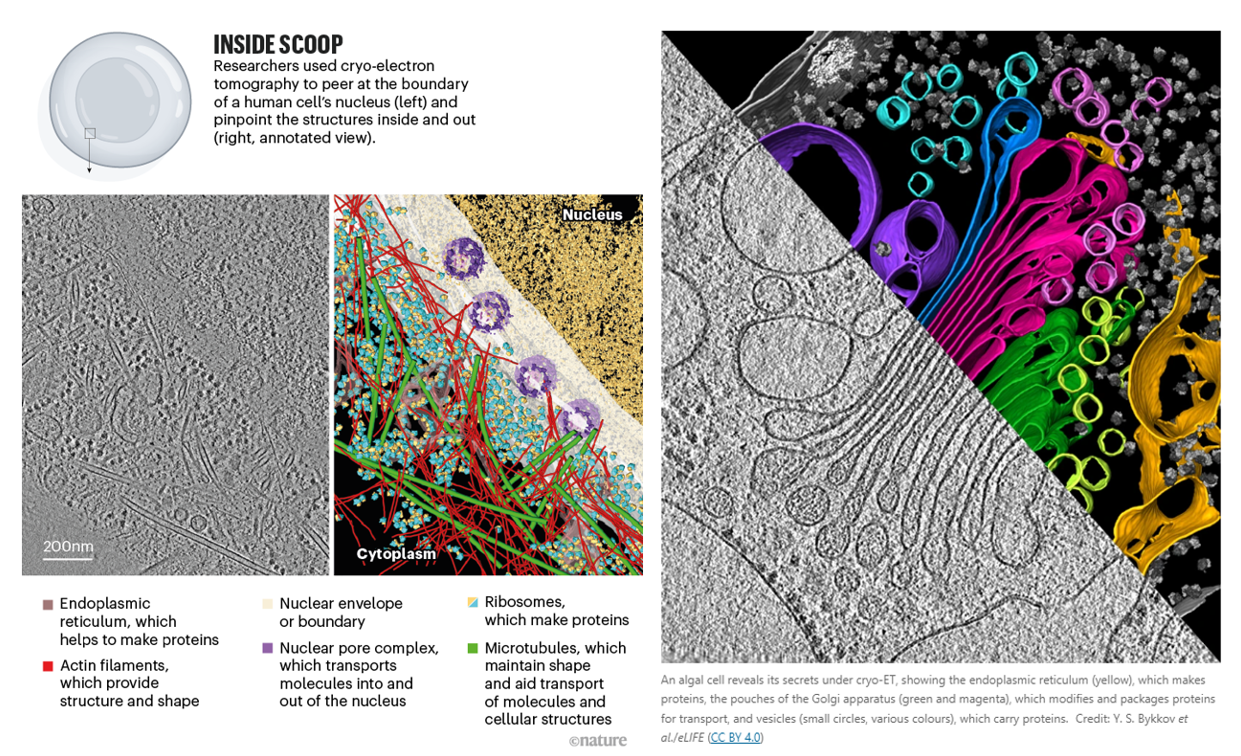
Because light couldn’t peer within cells until 15 years ago, researchers have for almost a century been using higher frequency particulars to study our molecular machinery. X-rays have wavelengths 1,000x than that of light and electrons 100,00x shorter, enabling visualization at the precision of individual atoms. Indeed, DNA’s structure “the double helix”, likely the most iconic visual in science, was discovered in 1953 by blasting x-rays at it and deciphering the diffraction pattern as the beams bounce off the structure. As seen below, our ability to control this process has progressed dramatically over time for both EM & ET, thanks to advances in the instrument’s hardware as well as the use of sophisticated aberration-correcting algorithms.
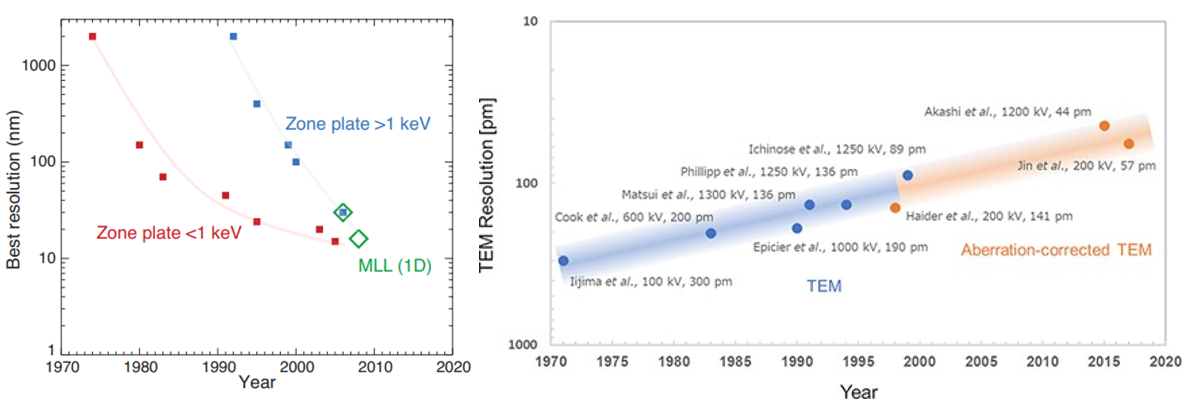
To be more specific, Rosalind Franklin’s DNA imaging used a technique called x-ray crystallography which turn molecules into crystals to stop them from wiggling and moving about. That’s no easy feat because these machines are built for action. Big, floppy objects tend to be more difficult to persuade to form crystals, so generally, the smaller and more rigid proteins are more amenable. Once crystalized, the “highly ordered lattice of tightly packed molecules” is then rotated through the x-ray beam. The resulting pattern of diffracted beams is transformed into a map of electron density, which the researchers combine with the protein sequence to build a 3D model. The technique has been optimized over the seven decades and continues to be the dominant method of determining molecular structure, contributing to 12 Nobel Prizes along the way. The method accounts for ~90% of the 200,000 proteins in the Protein Data Bank – the most popular public repository for such structures.

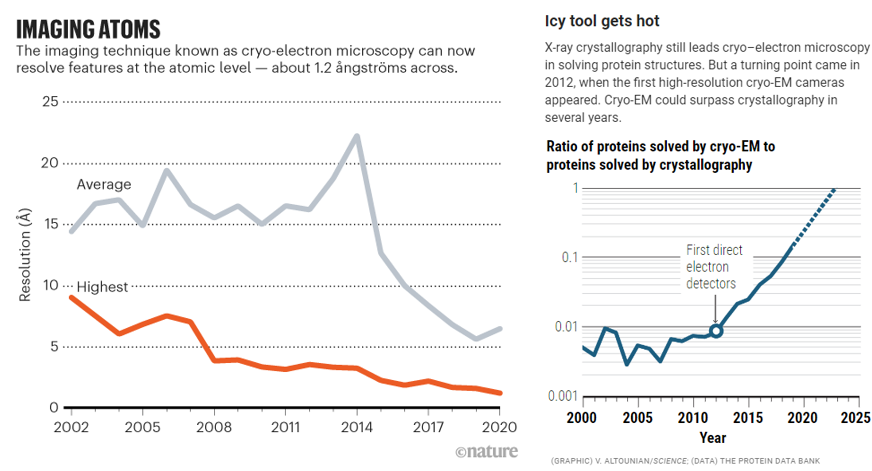
Since then, two new technological paradigms have swept through the structural biology field: cryo-electron microscopy (cryo-EM) and most recently cryo-electron tomography (cryo-ET). These pulse the objects with electrons instead of x-rays and contain their movement in different ways. Cryo-EM has distinct advantages over its predecessor but is a couple decades less mature and easily useable. And, cryo-ET improves upon its forefather but is several decades more nascent still.
Cryo-EM flash freezes individual molecules in a veil of water nanometers thick, allowing them to be captured in a freeze frame roughly as they are in the cell. In comparison, its predecessor stabilizing them by putting them in a chemical straitjacket so they can then be crystallized. This interference causes the scene to lose valuable information. The unnaturalness of the method makes it difficult to, for example, crystallize cellular membrane proteins, a drug designer’s best friend, because they constantly flex in and out of the cell.

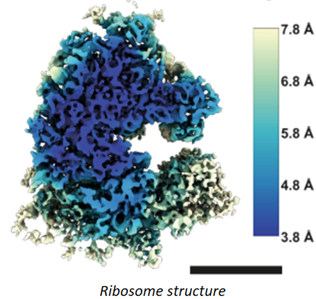
For years, researchers fretted that the technique couldn’t get precise enough. Before 2010, the field couldn’t break through the 0.4nms barrier – four times the width of a hydrogen atom, not good enough to measure individual amino acids. And that was the world’s best reporting; the field’s average measurement was on the scale of 1.5nms. In 2010, machines like Krios combined with the first direct electron detectors that track the electron’s paths before it fries the sample have flipped the script. Researchers have achieved 0.12nms, on par with crystallography’s precision. As its resolution has quickly approached that of an atom, the technology’s developers also improved its throughput: detectors could produce 6,000 images a day in 2020, several times the rate a few years prior. Hence, researchers have been switching over to it in droves. It’s now resolved the structure of 10,000+ biological molecules, and the EMDB has gone from 8 structures in 2002 to 1,470 today.
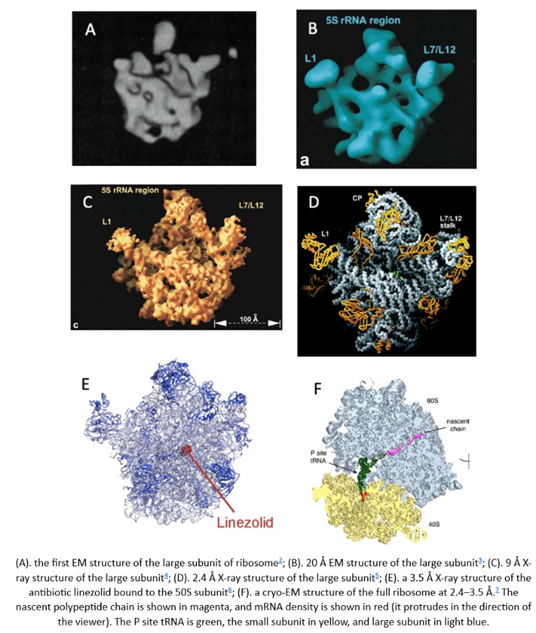
What does that improvement in resolution charted above actually look like in practice? The images below show cryo-EM photos over time from the first structure of the ribosome taken in 1984 (A) to one taken of the same structure in 2019 (F). The decades-long struggle towards atomic-level precision for a single, well understood, and abundant molecule should be underscored by the fact that it’s made of a quarter million atoms. That’s a lot of atoms to pinpoint in space. On the other hand, smaller and rarer molecules are harder to detect in the first place. Nothing comes easy at the scale of atoms.
Cryo-EM, like all technologies, has its limitations. For instance, resolution:
In cryo-EM, proteins and other macromolecular complexes are flash-frozen in a thin layer of water, ideally not much thicker than the protein itself. Irradiating that layer with low-energy electrons produces 2D images of individual particles on the detector — fuzzy shadows cast from scattered electrons (see ‘Modelling in ice’). Thousands or even hundreds of thousands of these noisy images are then computationally sorted and reconstructed to create a 3D map. Finally, other types of software fit the protein sequence into the map to create a model. The smaller the object, the noisier the images, so cryo-EM tends to work best for larger structures.
To avoid mistaking noise for signal, researchers typically split particles into two subsets and build ‘half maps’ from each. The correlation between those two maps is used to calculate resolution — but it’s an imperfect proxy. The resulting values must be taken with a grain of salt.
Additionally, some practices introduce unscientific bias, like creating a ‘‘mask’ of the expected overall shape of the protein and use that to exclude portions of images. Done judiciously, this boosts the signal-to-noise ratio; done aggressively, it shoehorns or ‘overfits’ data.
And don’t forget about cost. The Krios machines cost $7 million and the detectors, sold separately, go for $1 million a pop. Plus, it costs $10K a day to run the machine as it requires energy 200,000x that of the voltage of the common outlet. As of 2020, 130 Krios machines had been sold by Thermo Fisher Scientific and installed around the world. Given the price tag, the machines are scarce commodities, even at the snazziest laboratories. Cambridge’s prestigious LMB, home to a dozen Nobels, has the luxury of three for a relatively small staff, and yet even its researchers must wait a month or more to get time. Most structural biologists have no access at all. "The wait can be from 3 months to infinity. It's becoming the haves and the have-nots."
However, the pioneer of the cryo-EM machine, awarded the Nobel for his work, is trying to convince manufacturers to make a minimum viable version for $1 million, which would go a long way to democratizing the instrument. He built a prototype of the low energy instrument he envisions from spare parts. He used it to acquire five structures in one week, roughly what a commercial machine could do, with 0.34nm resolution. So far, at least one company has taken a stab at building them. He's since optimized the design of the low energy cryo-EM machine, developing one that costs up to 10x less than current high-end models.
In the future, further improvements may allow 0.1nm resolution, says one of the researchers that set the latest resolution record. But diminishing marginal returns set in fast beyond that. “All the chemistry stops at 0.1nm resolution, and the biology mostly stops at 0.3nm… Many drug candidates, however, are based on the chemistry of small molecules, not whole proteins. This is where sub-2 Å resolution could make a difference, allowing researchers to conduct structure-based design of better drugs.”
New techniques to for the first time study and visualize the molecular machines driving processes like DNA transcription:
In humans, some molecular motors march straight across muscle cells, causing them to contract. Others repair, replicate or transcribe DNA: These DNA-interacting motors can grab onto a double-stranded helix and climb from one base to the next, like walking up a spiral staircase. To see these mini machines in motion, the team wanted to take advantage of the twisting movement: First, they glued the DNA-interacting motor to a rigid support. Once pinned, the motor had to rotate the helix to get from one base to the next. So, if they could measure how the helix rotated, they could determine how the motor moved.
But researchers face a problem: Every time one motor moved across one base pair, the rotation shifted the DNA by a fraction of a nanometer. That shift was too small to resolve with even the most advanced light microscopes.
Two pens lying in the shape of helicopter propellers sparked a solution: A propeller fastened to the spinning DNA would move at the same speed as the helix and, therefore, the molecular motor. If they could build a DNA helicopter, just large enough to allow the swinging rotor blades to be visualized, they could capture the motor’s elusive movement on camera.
To build molecule-sized propellers, Kosuri, Altheimer and Zhuang decided to use DNA origami. Used to create art, deliver drugs to cells, study the immune system, and more, DNA origami involves manipulating strands to bind into beautiful, complicated shapes outside the traditional double-helix.
“If you have two complementary strands of DNA, they zip up,” Kosuri said. “That’s what they do.” When one strand is altered to complement a strand in a different helix, they can find each other and zip up instead, weaving new structures.
To construct their origami propellers, the team turned to Yin, a pioneer of origami technology. With guidance from him and his graduate student Dai, the team wove almost 200 individual pieces of DNA snippets into a propeller-like shape 160 nanometers in length. Then, they attached propellers to a regular double-helix and fed the other end to RecBCD, a molecular motor that unzips DNA. When the motor got to work, it spun the DNA, twisting the propeller like a corkscrew.
“No one had seen this protein actually rotate the DNA because it moves super-fast,” Kosuri said.
The motor can move across hundreds of bases in less than a second. But, with their origami propellers and a high-speed camera running at a thousand frames per second, the team could finally record the motor’s fast rotational movements.
“So many critical processes in the body involve interactions between proteins and DNA,” said Altheimer. Understanding how these proteins work—or fail to work—could help answer fundamental biological questions about human health and disease.
The team started to explore other types of DNA motors. One, RNA polymerase, moves along DNA to read and transcribe the genetic code into RNA. Inspired by previous research, the team theorized this motor might rotate DNA in 35-degree steps, corresponding to the angle between two neighboring nucleotide bases.
ORBIT proved them right: “For the first time, we’ve been able to see the single base pair rotations that underlie DNA transcription,” Kosuri said. Those rotational steps are, as predicted, around 35 degrees.
Millions of self-assembling DNA propellers can fit into just one microscope slide, which means the team can study hundreds or even thousands of them at once, using just one camera attached to one microscope. That way, they can compare and contrast how individual motors perform their work.
“There are no two enzymes that are identical,” Kosuri said. “It’s like a zoo.”
One motor protein might leap ahead while another momentarily scrambles backwards. Yet another might pause on one base for longer than any other. The team doesn’t yet know exactly why they move like they do. Armed with ORBIT, they soon might.
ORBIT could also inspire new nanotechnology designs powered with biological energy sources like ATP. “What we’ve made is a hybrid nanomachine that uses both designed components and natural biological motors,” Kosuri said. One day, such hybrid technology could be the literal foundation for biologically-inspired robots. (link)
- Subcellular Frame (10s to 100s nms)

The other major limitation of cryo-EM is that it can only examine isolated molecules. Some researchers are aiming to push the field being that limitation:
In an exciting emerging direction for cryo-EM, some methods for interpreting the heterogeneous structure data dare to go even further. These approaches aim to not only to sort static structure snapshots into discrete states, but also to extract information about dynamics, interpret how such states are connected, and infer how a structure continuously progresses from one conformation to the next. Such methods are based on algorithms including multi-body refinement (e.g. 1), manifold embedding (e.g. 1,2) and increasingly rely on the power of deep learning (e.g. 1,2,3). These advanced algorithms will allow researchers to generate high-resolution ‘molecular movies’ of macromolecules based not on conjecture but on actual experimental data, and will provide us with deeper insights into their functional motions and mechanisms. (link)
Another research group reported a method called cryoID9, which melds cryo-EM with various other techniques. One of these involves breaking open cells in a method that enables proteins to partially remain inside the original cellular milieu; in this way, researchers can view proteins in a near-native state. (link)
However, these adaptions underscore that cryo-EM was merely adopted to a multi-object view; cryo-ET was born in it. The successor to the throne, cryo-ET, can obtain information about molecular structures in their native environments at nanometer-scale. It thereby bridges the gap between structural biology and light microscopy. “It’s not quite structural biology, and it’s not quite cell biology — it’s sort of like some hybrid in between.” Another researcher, a biophysicist at the Max Plank Institute and a pioneer of cryo-ET, described it as “molecular sociology.” “It’s like having a photo of a whole crowd, rather than one person’s headshot. And this is how proteins live, after all. Proteins are social — at any given time a protein is in a complex with about ten other proteins.”
The technique involves freezing the whole cell, then using a focused ion beam to etch away at the top and bottom of the sample to make a thin slice called a lamella. It’s rotated around as researchers take snapshots of a chunk of a cell in its natural state, teaming with molecules, from a bunch of angles. It’s as difficult as it sounds, and has the been in the works for 40 years:
Getting the technique to deliver on its promise took decades of work, he says. “It was only less than 20 years ago that we could really do what we were hoping to do, which is look at the molecular architecture of cells.” One of the challenges was that streams of electrons are extremely damaging to biological samples, which made it difficult to capture enough snapshots for a clear, crisp image. Those decades have seen considerable methodological improvements, including the introduction of focused-ion-beam milling, ultrasensitive electron detectors and higher-quality microscopes. The focused ion beam technique allowed scientists to move from just prokaryotes (smaller and thinner) to eukaryotes as well.
Cryo-ET still has more than enough difficulties to work through before it challenges the other two techniques as the dominant paradigm. “Cryo-ET is where cryo-EM was in the early 90s, long before it was able to achieve atomic resolution,” says a biophysicist at UCLA.
It’s already been used to make meaningful progress on basic science and therapeutics:
Groups such as Baumeister’s have used this method to examine how proteins associated with neurodegenerative diseases such as Huntington’s6 and motor neuron disease (amyotrophic lateral sclerosis, or ALS)7 interact with components of the cell such as the endoplasmic reticulum (ER), a large piece of cellular machinery that helps to synthesize proteins. Researchers have found that neurotoxic clumps of proteins implicated in these diseases behave very differently to one another inside cells. In Huntington’s, for example, aggregates of a mutant form of a protein called huntingtin seem to throw the organization of the ER into disarray, whereas in ALS, aggregates of an abnormal protein impair the cell’s biochemistry by activating its protein-degrading machinery.
In the future, scientists hope to use such methods to better understand how therapeutics work, by visualizing how drugs act on the molecular innards of cells. In an early demonstration, Julia Mahamid at the European Molecular Biology Laboratory in Heidelberg, Germany, and her colleagues were able to glimpse antibiotics in a bacterial cell binding to ribosomes — organelles that serve as protein factories8. The feat was made possible by pushing the resolution of cryo-ET to 3.5 ångströms.
It was also used to visualize the ribosome translating DNA into RNA at atomic detail inside a bacteria:
Translation is the fundamental process of protein synthesis and is catalysed by the ribosome in all living cells1. Here we use advances in cryo-electron tomography and sub-tomogram analysis2,3 to visualize the structural dynamics of translation inside the bacterium Mycoplasma pneumoniae. To interpret the functional states in detail, we first obtain a high-resolution in-cell average map of all translating ribosomes and build an atomic model for the M.pneumoniae ribosome that reveals distinct extensions of ribosomal proteins. Classification then resolves 13 ribosome states that differ in their conformation and composition. These recapitulate major states that were previously resolved in vitro and reflect intermediates during active translation. On the basis of these states, we animate translation elongation inside native cells and show how antibiotics reshape the cellular translation landscapes. During translation elongation, ribosomes often assemble in defined three-dimensional arrangements to form polysomes4. By mapping the intracellular organization of translating ribosomes, we show that their association into polysomes involves a local coordination mechanism that is mediated by the ribosomal protein L9. We propose that an extended conformation of L9 within polysomes mitigates collisions to facilitate translation fidelity. Our work thus demonstrates the feasibility of visualizing molecular processes at atomic detail inside cells.
But it still faces a lot of issues to be worked through:
- Difficulty of searching or identifying the desired object:
Unlike the purified protein samples used for cryo-EM, tomography entails searching for biological structures in their natural environment — with an emphasis on the ‘search’. “The dirty secret of cryo-ET is that a tomogram covers like 0.001% of a mammalian cell,” says Villa. The odds are therefore stacked against any individual lamella containing the specific protein or event that a researcher is seeking. The chances are better with microbial specimens — Villa estimates that researchers can cover roughly 30–50% of a bacterial cell with a tomogram — and microbiology is a major focus for cryo-ET researchers.
Specialists such as Villa recommend researchers do their homework before starting out — using other methods to understand how abundant a protein is, or the factors that influence the frequency with which an event occurs, for instance.
Still, studies can easily become fishing expeditions. In 2022, cell biologist Benjamin completed a years-long effort to reconstruct the cellular machinery that the algae Chlamydomonas uses to assemble and maintain its cilia — hair-like structures involved in movement and environmental sensing2. “We were just milling these algae cells randomly, trying to hit this structure at the base of the cilium,” says Engel. “It took many years to gather this one structure.” Faster lamella-generation workflows are making this effort less painful, and Zhang says that automation has increased the number of samples that can be prepared from 5 or 6 per day to 40. Still, solving a structure can require hundreds of samples.
Researchers can also narrow down the cellular terrain that they need to comb using a technique known as correlative light and electron microscopy (CLEM). With this, samples are fluorescently labelled to mark specific proteins or compartments in the cell in which an event of interest is most likely to occur prior to freezing. The sample is then imaged under a specially designed cryo-fluorescent microscope so that researchers can better target lamellae.
In 2020, for instance, Villa and her colleagues used CLEM to reveal how a mutated protein associated with Parkinson’s disease interferes with the trafficking of essential biomolecules between different parts of the cell3. “Only 30% of the cells even had the phenotype that we were looking for, and if we had just done random lamellae in random places, we would’ve never found it,” says Villa.
But as with all things cryo-ET, aligning fluorescence and electron-microscopy data isn’t straightforward. The resolution produced by standard fluorescence imaging is so much lower than in electron microscopy that producing optimal lamellae can still require a lucky break. “Sometimes your whole image at electron-microscopy magnification would be just one pixel in the light microscope,” explains structural biologist Kay Grünewald at the University of Oxford. Groups are working to merge CLEM with single-molecule-resolution fluorescence microscopy, but that remains a work in progress. For now, Grünewald and others often use tiny fluorescent beads that are electron-dense enough to be detected with an electron microscope, making it easier to triangulate locations between the two imaging systems.
But moving samples between multiple instruments creates its own difficulties, including increased opportunity for contamination. And Julia Mahamid, a structural biologist at the European Molecular Biology Laboratory in Heidelberg, Germany, notes that even a frozen sample can warp and shift during the preparation process. Without fluorescent imaging at this stage, these perturbations might go unnoticed.
A new generation of integrated cryo-CLEM systems promises to ease these challenges. The ELI-TriScope platform4, for instance, developed at the Chinese Academy of Sciences, Beijing, provides researchers with colocalized electron (E), light (L) and ion (I) beams. Structural biologist Fei Sun, one of the lead researchers on this effort, says that the system has taken a lot of the guesswork out of his study of the centrioles that sort and separate chromosomes during cell division.
Meanwhile, because electron microscopy is missing an equivalent of green fluorescent protein — in which a genetically encoded reporter can be used for cellular labelling in fluorescence-imaging experiments — the hunt is under way for labelling methods that would allow researchers to home in on targets of interest without fluorescence imaging. Existing options, such as gold nanoparticles coupled to protein-binding functional groups, are too bulky and have the potential to artificially aggregate many targets at once. Baumeister is concerned that such tags could perturb the nanometre-scale details that cryo-ET is used to uncover. Nevertheless, some have seen promising results. For example, Grünewald and his colleagues have developed DNA-based ‘origami’ tags that fold into electron-dense asymmetrical shapes that bind and point towards cellular features of interest. Although the tags are currently restricted to external proteins, Grünewald is exploring ways to deliver them into the cell itself. “It’s not as versatile yet as we hope, but for certain questions it works quite well,” he says. (link)
- Resolution:
Unfortunately, even high-quality raw cryo-ET data resemble the grainy monochrome static of an untuned analogue television. Seasoned veterans can discern features such as lipid membranes or mitochondria, but finding individual proteins or complexes is a trickier proposition.
The standard approach is to use template-matching software, which exploits existing structural data to pick out the target amid the whorls of black and grey. Although relatively fast, this approach can be sloppy, and Mahamid points out that even well-studied molecular assemblies such as the protein-synthesizing ribosome can slip through the net. The process is even less reliable for small, less abundant or disordered proteins — and, by definition, requires prior structural knowledge about the target. (link)
- Cost: More trained personnel with access to the highly specialized — and costly — equipment are also needed. Baumeister estimates that developing a full workflow for cryo-ET can cost upwards of €10 million (US$10.7 million). “That’s much more than the start-up package of a typical young faculty member,” he says. (link)
- Sample thickness: limited to ~500 nm, making many bacterial, most archeal, and nearly all eukaryotic samples unamenable to imaging. (link)
AI / ML may significantly improve the searching problem:
Deep learning is also helping researchers to annotate their tomograms in a more automated fashion. But these tools, too, often use templates, including the DeePiCt algorithm7, developed by Mahamid and her colleagues. Mahamid describes it as a vast improvement over earlier template matching, but adds that “we’re still missing about 20% of our particles”.
For now, what the cryo-ET field most urgently needs is data. Artificial intelligence algorithms are only as good as their training sets, and progress in deep learning for image processing and analysis will require a vast repository of annotated images that can guide the picking and interpretation of individual particles from the complex cellular soup. (link)
CZI is helping collect data, but it’s database only contains 120 structures so far.
Other researchers are combining cryo-ET with another technique called X-ray tomography that can capture images of whole cells. This allows scientists to examine the structure of larger components, such as mitochondria or nuclei, and then zoom in on specific areas of interest.
Some labs have already accomplished this feat. Maria Harkiolaki, principal beam-line scientist at the Diamond Light Source, a synchrotron facility in Didcot, UK, and her colleagues recently published12 a model of the mechanics of SARS-CoV-2 infection that uses cryo-ET and X-ray tomography to elucidate the process. They captured events at both the level of the cell and individual molecules, and proposed an idea for how the virus replicates in primate cells. (link)
There are still other cryo techniques being developed, like the Franklin Rosalind Institute’s (FRI) cryo-plasma seeking to make “mesoscopic maps of whole cells and tissues in pristine detail.” Here’s a picture:

Many techniques founded well outside of the structural biology world are applied to subcellular phenomena. Some are working to record cells over time:
- FRI made the world’s first time-domain electron microscope for life science: “The microscope, known as ‘Ruska’, will work with biological samples both cryogenically frozen and in liquids, which will enable imaging of molecules in motion. By combining high speeds with liquid cells, the Ruska microscope will be able to ‘film’ proteins as they fold or image drugs interacting with other molecules. For cryogenically frozen samples, the frames captured as the beam passes through the sample will enable the creation of 3D models of biological structures, such as viruses or proteins.”
- Another device facilitates the video recording of subcellular action over a defined period of time. “Every single cell is tracked from beginning to end… This not only allows you to analyze a lot of different parameters at once, even with a few cells, but also if you’re interested in using this as quality control for your T cell product, it’s probably as rigorous as you can get.”
- Nucleic acids (DNA and RNA) are amplified easily and hence single cell technologies quantifying nucleic acids have been developed rather quickly. By contrast, understanding the dynamics of cells and how they move, change, adapt and evolve to their microenvironment — and how they interact with other cells — is very difficult to study. My lab, very early on, recognized the importance of dynamics. Cells are constantly changing and it is their functional capacity, rather than the abundance of nucleic acids or proteins that defines them. Our TIMING (Time-lapse Imaging Microscopy in Nanowell Grids) platform parallelizes thousands of single-cell interactions such that scientists are now able to watch each cell live its own life.
Others only in the exact moment of interest, like when a nerve cell fires:
Physicist Ilaria Testa at the KTH Royal Institute of Technology in Stockholm built a smart microscope to observe subcellular vesicles as they release calcium at neural synapses when nerve cells fire — a key step in signal transmission. “These are rare events and it’s not always easy to capture them.”
One option was to image a specimen continually in the hope of capturing the moment. But vesicle-release events are transient and the structures too small for standard microscopy. Super-resolution imaging can reveal more detail, but it requires high-intensity light sources that can be used only briefly before the sample is damaged. The team tried various time-lapse approaches, in which images are captured at regular intervals. But that, Testa says, was like watching a soccer match and missing a goal because you are looking elsewhere at the crucial moment. “It was a bit frustrating,” she says. “The resolution would have been fine if we only knew where to look.”
Testa and her team combined two microscopy approaches: a fluorescent wide-field microscope and a form of super-resolution microscopy known as stimulated emission depletion (STED). They developed a software system to control these microscopy modes: when the software detected a change in fluorescence, the system would switch automatically to the higher-resolution STED mode. This allowed the team to capture — with nanometer precision — how cells reorganize their synaptic vesicles after releasing calcium3.
Testa and her team took about two years to build the microscope, she says, using mostly custom 3D-printed hardware. The set-up cost about $200,000: most expensive were the lasers.
Still other groups are looking to make super-resolution light microscopy (i.e. those with resolution below Abbe’s limit of 200nm for light) cheaper and more accessible. One group’s using a computational approach:
In 2016, Henriques and his team devised a computational method called super-resolution radial fluctuations (SRRF) to extend super-resolution microscopy to conventional fluorescence microscopes, which are generally widely accessible4. The method now relies on a machine-learning approach that adapts the microscope to acquire images at high speeds and then parse the data to identify sample-specific blips in fluorescence. Users capture 100 images of their sample at high speeds to minimize chemical damage to fluorescent labels, known as bleaching, as well as to reduce blurring from movements. The data are then fed into an algorithm that looks for variations in fluorescence.
The fluorescent molecules, or fluorophores, that are used to label cellular structures fluctuate in brightness depending on their location and other sample properties. “Every time [the algorithm] picks up a fluctuation, it’s able to pinpoint the exact center of the fluctuation and if it’s coming from a fluorophore or not. So, it generates something that’s crisper than the original.
Another’s applying the DNA nanotechnology techniques it helped pioneer:
Key to the Wyss Institute’s DNA-driven imaging super resolution technology is the interaction of two short strands of DNA, one called the “docking strand” that is attached to the molecular target to be visualized and the other, called the “imager strand”, which carries a light-emitting dye.
Despite the recent revolution of optical imaging technologies that has enabled the distinction of molecular targets residing less than 200 nanometers apart from each other, modern super-resolution techniques have still been unable to accurately and precisely count the number of biomolecules at cellular locations. With DNA-PAINT super-resolution microscopy and qPAINT analysis, researchers will be able to quantify the number of molecules at specific locations in the cell without the need to spatially resolve them, without requiring an expensive large super-resolution microscope.
Ultivue, Inc., a startup launched out of the Wyss Institute, is focused on developing imaging solutions to unfold new areas of biology by leveraging DNA-PAINT to enable the precise co-localization of an unlimited number of targets.
- Intercellular Frame (100s to 1,000s nm)
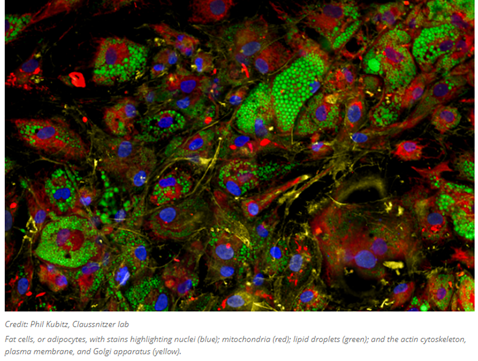
The high-throughput quantitative imaging tool pictured above is the very latest method helping to bridge the gap between shape and function. The image analysis software gathers data on over 3,000 cellular attributes, offering a comprehensive perspective on how genetic variations, small molecule or genetic interventions, and other factors affect the shape, composition, and functionality of cells. Whereas prior iterations of similar techniques identify eight general organelles or cellular components, this one employs six fluorescent dyes that collectively highlight the structure and behavior of nine distinct cellular compartments: the nucleus, nucleoli, mitochondria, endoplasmic reticulum, Golgi apparatus, cytoplasmic RNA, f-actin cytoskeleton, plasma membrane, and lipid droplets (organelles responsible for lipid regulation present in various cell types, especially adipocytes). It then obtains image-based features from stained cells and generates cellular profiles, classifying cells based on subtle patterns that are often imperceptible to the human eye.
The lab of the tool’s inventor is now strengthening the machine-learning capabilities to track information flow within single cells from transcription to phenotypes. Experimentally, the lab uses the tool to “assess the relationships between genetic variants and cellular programs, to understand highly granular processes of disease biology. These patterns provide direct insights into the field of disease biology… It moves the community from a very targeted, hypothesis-driven, unidimensional cell-based assay framework to one that allows us to survey biological processes in a cell and assign function to genes and genetic variations in an unbiased manner.”
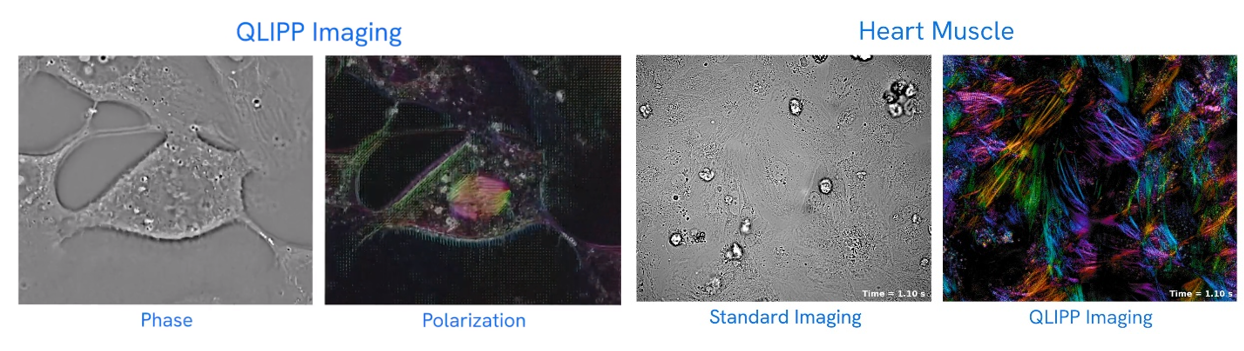
Other devices avoid dying, staining or labeling the cells by analyzing the way light refracts through the sample:
To understand how it works, “think of straw in a clear glass of lemonade.” In the air, the straw appears normal, but in the lemonade, it appears bent, or refracted. In a cell, each organelle refracts light differently, as if each organelle were a different lemonade. CX-A accounts for the variations in refraction and reconstructs the three-dimensional image formed by the interference of the refraction of the cellular components in its field of view.
The company’s new technology builds on prior design but is automated so scientists can program the various fields of view they would like to observe in a single slide or in 96-well plates, then walk away and let the machine do the work. The tool is a tenth of the price of a super-resolution confocal microscope, which typically runs between $300,000 and $500,000.
The group used the instrument to observe mitochondrial fission and fusion, the way groups of cells interact and react to one another, and other cellular phenomena.
CZI developed a technology that also enables the high resolution of cells without the need for stains or labels. It utilizes two properties of light: phase and polarization that are also altered in specific ways as it moves through translucent materials. The software works with inexpensive hardware and microscopes common to most labs. They made it freely available.
Tissue-level frame (1,000s of nm’s)
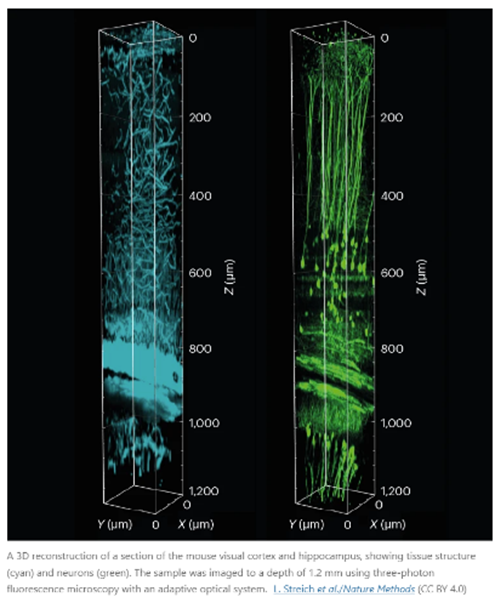
While achieving millimeter resolution is of course no challenge, imaging live organisms is exceptionally difficult. Not only does the tissue quite quickly block light from penetrating the skin but the entire structure is moving with every heartbeat, with every inhalation, and with every muscle twitch. Advanced computational techniques are helping circumvent these issues. Consider these two stories:
A mouse’s heart beats roughly 600 times each minute. With every beat, blood pumping through vessels jiggles the brain and other organs. That motion doesn’t trouble the mouse, but it does pose a challenge for physicist Robert Prevedel.
Prevedel designs and builds microscopes to solve research problems at the European Molecular Biology Laboratory in Heidelberg, Germany. In his case, the issue is how to capture neural activity deep inside the brain when the organ itself is moving. “The deeper we image, the more aberrations we accumulate,” he says. “So, we have to build our microscope to quickly measure and adapt.”
Typically, microscopes struggle to peer deeper than about one millimetre into tissues. Beyond that, light bouncing off intracellular structures creates distortions, blurring the images — even when the sample is not moving to a heartbeat.
In 2021, Prevedel and his colleagues engineered a microscope that combines an approach called three-photon fluorescence imaging, which is used to probe inside tissues, with an adaptive optical system. The latter strategy was first developed in astronomy and directs light using mirrors and lenses made of deformable membranes instead of rigid optical materials. Software tools quickly alter the membranes’ shapes in response to variations in the sample, bending light in specific ways to peek through surface structures. Rather than needing a human operator, this ‘sample-adaptive’ approach relies on the microscope itself adjusting the optics in real time to continually produce high-quality images.
Prevedel’s microscope had to go one step further to compensate for the vibrations of the animal’s pumping heart. By timing photos to the heartbeat, the team was able to image cells nearly 1.5 millimetres beneath the brain’s surface in a region called the hippocampus, and 0.5 mm deeper than previous efforts1. “It was really exciting to see that it worked much farther down than we were expecting,” Prevedel says.
Prevedel’s microscope is just one of a suite of smart microscopes that rely on adaptive optical systems such as deformable membranes, coupled with machine-learning approaches, to peer deeper into tissues than ever before, and to zoom in at crucial moments to capture fleeting moments in the life of a cell.
Often involving custom-built hardware and software components that can make them inaccessible to the broader biological community, these efforts are producing tools that do more than just produce clear images. They’re also gentler on living systems, allowing researchers to observe prolonged processes without damaging their samples, thus expanding the reach of microscopy.
Another group developing adaptive microscopes designed one to study how clumps of cells form complex tissues as the mouse embryo develops2. Led by developmental biologist Philipp Keller, the team wanted to image the developing embryo over three days, during which its diameter grows from about 200 micrometres to nearly 3 millimetres. Anchored on one end, the embryo is “free to sway in the breeze — it moves”, and its density and other optical properties change over time. “The microscope needed to keep up with all of that.”
She and her colleagues started with a technique known as simultaneous multiview light-sheet microscopy, which Keller’s team had developed to track cell movement in developing fruit-fly embryos. To adapt it for the mouse, they altered the optical design and built software to control such factors as the angle of the light source and the positions of optical elements. The software was able to gauge the data quality of the images as they were collected, and could tweak these factors to optimize images throughout the experiment.
The microscope was also automated to detect the growing embryo’s position in the sample chamber and keep it centred in the field of view, adjusting the distances to ensure consistent image quality. “A lot of biological specimens like to roam around, so you need to keep them centred,” McDole says.
McDole and her team used this system to observe mouse embryo development over a 48-hour period, imaging the embryonic heart and other developing organs at single-cell resolution. They have subsequently imaged brain organoids for up to two weeks. “That’s kind of the future of autonomous microscopy — letting the microscope decide when and where and how to act on specific events,” McDole says. “You can teach the microscope: ‘It’s going to be 3:00 a.m. when this cell division happens, and I want you to zoom in and do something and then go back to normal imaging.’”
The Keller group has released schematics for building such an instrument, and the software is freely available online. Researchers who already use a light-sheet microscope can expect to spend about US$30,000 to add these capabilities to their system, Keller says. “It’s a pretty small extra investment” to achieve deep-tissue, single-cell resolution, he notes.
- Organ-level Frame (1,000s of nm’s)
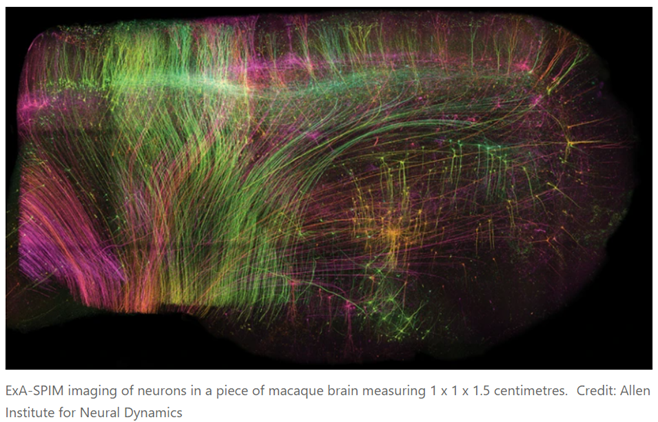
Researchers are increasingly relieved of having to choose between field of view and resolution. For instance, a new technique can image the entire mouse brain in under a day at nearly cellular resolution:
The mammalian brain is a multiscale system. Neuronal circuitry forms an information superhighway, with some projections potentially stretching dozens of centimeters inside the brain. But those projections are also just a few hundreds of nanometers thick — about one one-thousandth the width of a human hair.
Understanding how the brain encodes and transmits signals requires the alignment of events at both these scales. Conventionally, brain researchers have tackled this using a multi-step process: slice tissue into thin sections, image each section at high resolution, piece the layers back together and reconstruct the paths of individual neurons.
Zhuhao Wu, a neuroscientist at Weill Cornell Medicine in New York City, describes this last step as “like tracing a telephone wire in Manhattan”. In fact, he adds, it’s “even more complicated, since every neuron makes thousands, if not tens of thousands, of connections”.
Microscope makers have sought to allow researchers to take a wide-angled peek at a large chunk of tissue and still see the details up close, without having to first slice up the tissue and then reconstruct the axons across different sections. The challenge is that microscope objectives are generally designed in such a way that it is difficult to take high-resolution images of large samples.
A preprint published in June offers a solution1. First, the researchers chemically removed the lipids to make the tissue transparent. Then, they embedded it in a material called a hydrogel, which absorbs water, to expand the tissue to three times its original volume. Finally, they scanned it with a lens borrowed from a completely different field of science. In this way, it was possible to image whole mouse brains without the need for any slicing, and at a resolution of about 300 nm in the imaging plane and 800 nm axially (perpendicular to the plane), comparable to that of confocal microscopy, a technique widely used for high-resolution brain imaging. Called ExA-SPIM (expansion assisted selective plane illumination microscopy), the protocol was also used to image neurons in macaque and human brains.
The approach can image an entire mouse brain in under a day — much faster and at higher resolution than is possible with other whole-brain approaches that have been applied to axon projections, such as MouseLight and fMost, which use tomography, says Chandrashekar. And the fact that images require only limited computational reconstruction significantly increases the accuracy of the resulting data.
Wu notes that none of the system’s components is new — they are just assembled in a synergetic way. “It’s not the first attempt to do this but it is probably the best attempt that we have now,” he says.
The team has so far imaged 25 or so mouse brains, producing some 2.5 petabytes of data, which the team compresses five-fold and stores in the cloud. For data analysis, the researchers collaborate with Google, which provides machine-learning algorithms for processing the data and reconstructing images of the neurons.
That said, the microscope design is open-source and instructions for building it are available on GitHub. But Glaser says that the current version is a prototype that he plans to streamline and document over the next year.
Read next section: diagnostics and continuous health monitoring