Reading, Writing, and Editing Life’s Code (The Century of Biology: Part I.I)
- Reading the Code of Life: DNA & RNA sequencing
A beautiful aspect of biology is that all life is written in language like a novelist pens a book or a computer scientist codes a program. It has structure not unlike a sentence has syntax or a function has relational dependencies. It has deterministic outcomes: a certain sequence of letters instructs the organism to make a certain type of protein. And, we knew it was possible, at least in principle, to read that code ourselves, because nature can do it with ease. In fact, it’s really freaking good at transcribing its own code – DNA replication errors happen roughly one in every hundred million copies. However, it wasn’t until the last couple decades that we could do it at all.
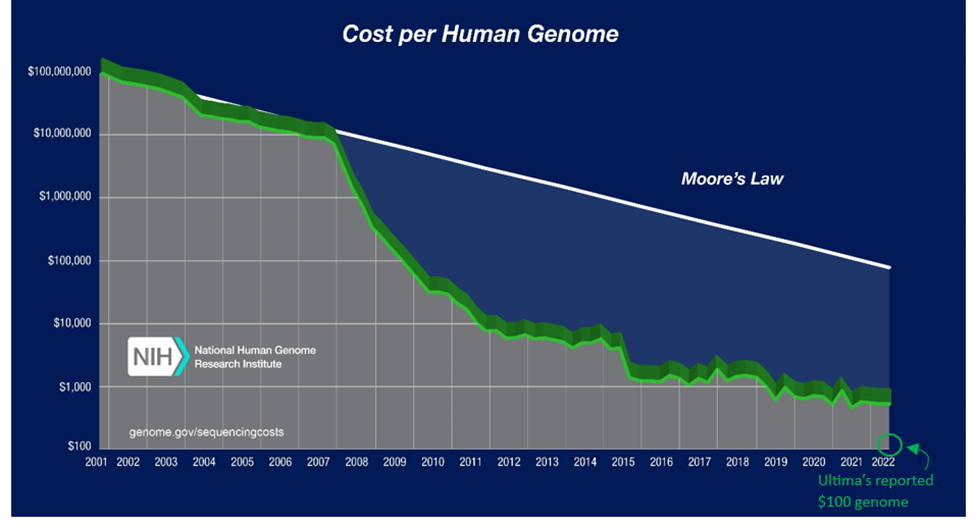
Sanger invented the first technique to reliably and reproducibly read life’s code in 1977. A decade later, the first commercial product released. In the early 2000s, a project costing $3B in government funding and 15 years enabled us to read the entirety of our own genome for the first time. Scientific and technological progress since then has been otherworldly. The cost to sequence our genome has fallen by one-million-fold in 20 years, outpacing Moore’s Law by six orders of magnitude. We can now do it for $100s and in as few as five hours.
Until the last decade, all DNA sequencing techniques have involved taking a bunch of identical genomes, cutting them up into tons of tiny pieces, and then using computational methods to try to put the pieces back together to the full length genome. It’s like taking a thousand copies of Macbeth, throwing them in a wood chipper, and then seeing if algorithms can figure out how Shakespeare did it.
The first successful attempts at reconstructing the human genome were so expensive in part because the technology at the time only allowed for roughly 50 base pair (bp) snippets. So, back of the napkin math would tell you they had to shred the three billion nucleotide bases into ~6 billion snippets. The trouble doesn’t stop there. Many sections of the genome have stretches where the same letters are repeated over and over again. Some have such stretches lasting tens of thousands of bases. The length of your sequencing technology’s snippets must be at least as long as these repetitive sections or else you end up hopelessly combing through thousands of identical snippets. For this reason, two decades after the Human Genome Project declared victory, 8% of the human genome still had never been successfully sequenced in full. In 2022, the Y chromosome, the most repetitive of our chromosomes and the remaining holdout, was successfully sequenced for the first time.
25-year-old Illumina has been the workhouse of the genomics revolution. It effectively used Sanger’s approach, but instead of doing one fragment at a time, it did millions simultaneously per run. That’s how you get million-fold cost cuts. Even according to Illumina, “The critical difference between Sanger sequencing and NGS is sequencing volume.” In addition to massively parallelizing the process, it massively refined its accuracy, with its error rates now less than 1 in 1,000bps.
While it remains the dominant player in the sequencing industry controlling 80% market share and generating 90% of the world’s sequencing data, its core products still primarily rely on short-read sequencing (SRS) approaches. This sequencing-by-synthesis approach falls prey to inversely compounded interest: it gets exponentially less accurate as read lengths increase. As such, researchers typically use it for reads under 200bps. Its highly accurate, easy-to-use, high-throughput, cheap SRS machines are great for a variety of applications like transcriptome analysis and targeted sequencing.
However, Illumina’s key patents expired last year, helping erode its pricing power over competitors. Far more importantly, several new companies have broken through with completely different approaches to sequencing. They introduced a new paradigm: long-read sequencing (LRS). In 2011, PacBio launched its enzyme-based technique, inspired by nature’s transcription method. Several years later, Oxford Nanopore came forward with a process that pulls the DNA through a cell membrane hole not much wider than the DNA itself, shoots electricity across the membrane as it does so, and records the voltage change as it refracts off the nucleotides.
Today, the market can be broken into three approaches to sequencing:
Flow-cell-based sequencers (e.g., Illumina sequencers) arrange monoclonal clusters of ~300-letter DNA fragments on a solid support tightly arrayed with nanoscopic landing pads. Under a camera’s watchful eye, a coordinated series of liquid washes containing dye-labeled DNA bases and custom-engineered enzymes will bind, measure, cleave, and extend the myriad DNA clusters. Onboard software algorithms watch each cycle, converting light signals into DNA base calls while being careful to filter out impure, noisy clusters from the final readout.
Semiconductor-based sequencers (e.g., PacBio sequencers) carefully load ~20,000-letter DNA circles into millions of zeptoliter (10-21 L) confinement chambers nano-etched into a metal substrate. Smaller than the wavelength of incident light, these so-called zero-mode waveguide (ZMW) chambers allow a camera to watch in real-time as highly-engineered polymerase enzymes incorporate dye-labeled DNA bases into the template DNA circles. Uniquely, these enzymes are armed with “photoshields”—chemical structures that shade fragile DNA molecules from the prolonged assault of laser light necessary to read off tens of thousands of bases.
Finally, the camera passes a tensor object to a transformer algorithm, converting twinkling light pulses into sequence data.
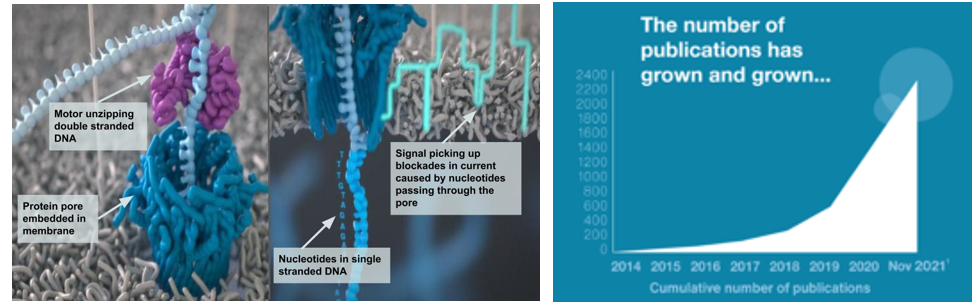
Nanopore-based sequencers (e.g., Oxford Nanopore sequencers) don’t use cameras. Instead, ~100,000-letter DNA fragments are unzipped by helicase enzymes before being ratcheted through an array of biological nanopores embedded in a membrane support. As each stretch of nucleotides passes through a pore, it creates a characteristic electronic trace (a squigglegram) that is translated by a deep-learning algorithm into sequence information. (Source)
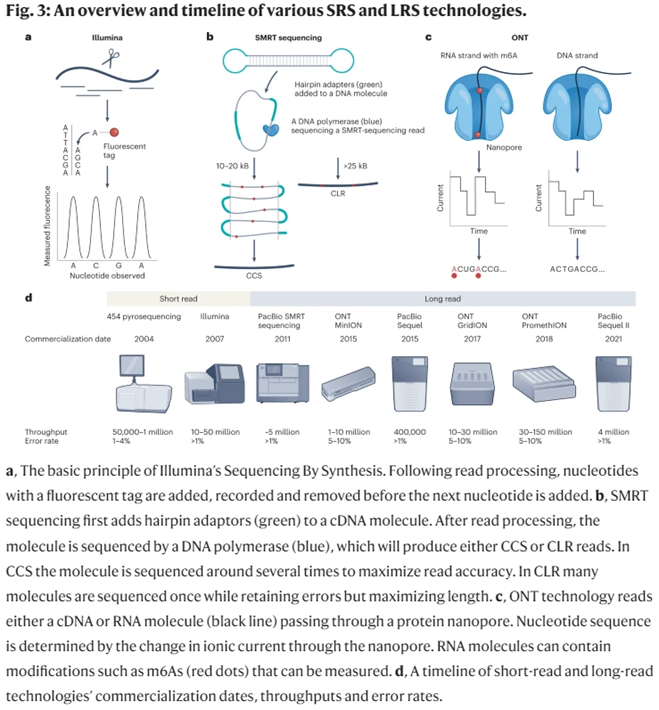
PacBio’s machines are optimal for highly accurate long reads. Their average continuous read length is ~15,000bps with error rates around 1%. Meanwhile, Oxford’s machines have been used to sequence DNA fragments up to 4 million bps long. It’s error rates are <5%. They also cost $500 and are the size of USB thumb drives, making sequencing accessible to anyone anywhere.
A brand new competitor, Ultima Genomics, made a jaw dropping entry by announcing last May that it had developed a machine to sequence a human genome for $100. It boasts longer read length than Illumina machines (∼300bp) and fast runs times (<20hrs) with high base accuracy (Q30 > 85%), at a low cost of $1/Gb. Read the rest of the pre-print for details on how it works, but here’s a summary:
Lauer attributes many benefits of the UG100 to a unique feature: a circular, open flow cell. Reagents are applied directly to a spinning silicon wafer that distributes them more efficiently than reagents pumped through a traditional flow cell, Lauer explains. In addition, the revolving design increases the speed of data collection and imaging, enabling Ultima’s sequencer to complete one run in about 20 hours, which he says is about twice as fast as existing technologies. (Source)
So far, only selected early access customers amongst the largest scale genome sequencing like Broad Institute and NY Genome Center have been given the chance to play around with the instrument. Initial reviews suggest it’s the real deal:
- Data from over 200 whole human genomes generated at the Broad Institute
- Published in Cell, a four million cell genome-wide PerturbSeq study demonstrates immediate usability for Ultima's platform in large-scale single-cell studies. The researchers found Ultima’s platform to be lower-cost, ultra-high throughput with equivalent results to Illumina’s.
- Whole genome methylation landscape of pre-cancerous tissues (pre-print)
- Multiple demonstrations of the ability to quantify circulating tumor by deep whole genome sequencing of cell-free DNA
So as not to see its $20B empire collapse in on itself, Illumina’s hot on the trail. It announced a new device that could parse the genome for $200. At over twice as fast as the prior model, it can churn out 20,000 whole genomes a year. Element also advertises $200. Remember again that the first time we did it, it cost $3 billion and 15 years. That was just 20 years ago.
Some other exciting new companies are Singular Genomics, Element Biosciences, and MGI.
- Writing the Code of Life: Synthesis
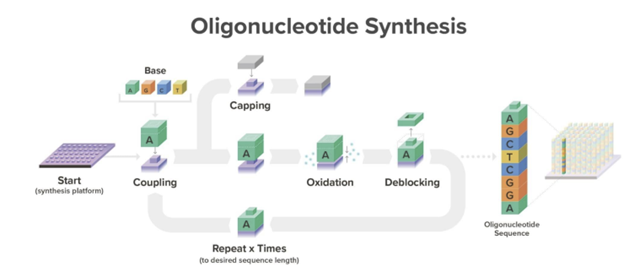
DNA chains are typically synthesized using a multi-step method known as phosphoramidite chemistry. In this procedure, a DNA strand is elongated by adding DNA bases one after the other. Each DNA base is equipped with a blocking group that prevents repeated additions of the base to the extending DNA chain. After attaching a blocked base to the DNA chain, acid is introduced to remove the blocking group and prepare the DNA chain for the subsequent base addition.
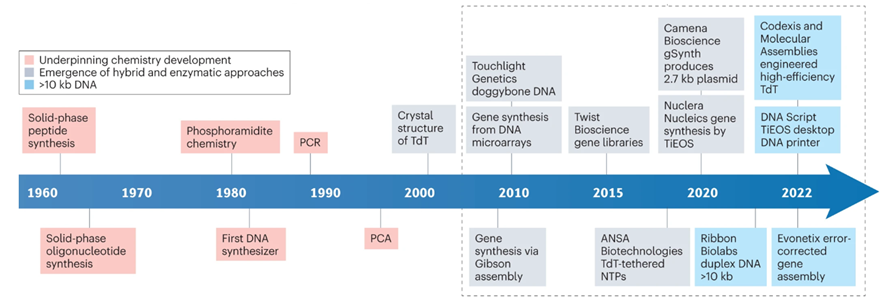
A tricky aspect of DNA synthesis is that it falls prey to negative compounding, limiting accurate synthesis lengths to a couple hundred bases. Every base added to the chain carries with it a failure rate. So, making a 200-base chain with a method that’s 99.9% accurate per block only yields 0.999^200 = 82% accuracy. A Again, nature’s failure rate is about one in a million bases so there’s plenty of room for improvement, but it’ll be a while until we’re cranking out entire genes in one go. Note that the limitation on our limited ability to synthesize long chains in continuous runs doesn’t cap our ability to make those long chains. We simply stitch the shorter chains together via subsequent rounds of PCR, ligation, and/or Gibson assembly. Pooled reactions are particularly efficient, allowing the assembly of 1 megabase fragments in 80 minutes.
Here’s a more detailed overview how the current method works[1]:
The DNA synthesis market first took off when Twist was able to miniaturize the phosphoramidite chemistry on a silicon wafer (chronicled here). Twist’s current technology allows for the parallel synthesis of 9,600+ strands of DNA. Twist’s success hinges upon parallelization which drives down cost, improves quality, and increases length of constructs (and turnaround time if you’re a DNA service company). However, phosphoramidite chemistry is still limited in sequence length (200-mers), accuracy and yield, due to imperfect nucleotide coupling efficiency, imperfect protection/deprotection chemistry, and depurination occurring as a natural side reaction. That being said, there are depurination-resistant methods and reagents which could theoretically allow for longer sequence lengths with phosphoramidite chemistry.
Pitfalls and Roadblocks in Chemical Synthesis
Although there have been major improvements in the world oligo synthesis and phosphoramidite chemistry, there are several large roadblocks preventing affordable and sustainable longer oligios and DNA. Fundamentally, chemistry is imperfect and 100% coupling efficiency is not possible. Even with extreme care, some oligos will not couple with the newly introduced nucleotide - causing short, prematurely truncated oligos. Another potentially detrimental scenario centers around improper protecting/deprotecting of the 5′ hydroxyl group on the sugar portion of the nucleotide. If this hydroxyl is not protected, an oligo with a “deletion” in its sequence can be produced (i.e. N-1 sized oligo). Additionally, larger oligos can undergo a common side reaction found in phosphoramidite chemistry that also wreaks havoc on oligos: depurination. Depurination, the loss of purine bases (adenine and guanine) from a nucleotide, creates an abasic site. This abasic site then reacts with the final alkaline nucleobase deprotection reagents and then also leads to prematurely truncated oligos forming (do you see a trend here?). In addition, phosphoramidite chemistry fundamentally uses large amounts of flammable organic solvents, like acetonitrile, which incurs high cost, creates potentially hazardous situations, and is largely harmful to the planet. These challenges have led other labs to look beyond chemistry for solutions.
The next-gen synthesis method is expected to be enzymatic[2]:
Given the pitfalls of traditional chemical synthesis, enzymatic methods of DNA synthesis have been proposed as an alternative to phosphoramidite chemistry. The most common form of enzymatic DNA synthesis is terminal deoxynucleotidyl transferase (TdT), used in nature to add bases to B and T-cell receptor sequences. Like phosphoramidite chemistry, TdT requires sequential and controlled addition of nucleotide bases but has not reached the n-mer length of phosphoramadite synthesis. To add to this, there can also be complications with secondary structure of oligonucleotides but this can be controlled by increasing temperature to prevent secondary structure annealing. Secondary structure is harder to control with enzymatic synthesis because higher temperatures can decrease polymerization rate, or even lead to protein denaturation, which is not a problem in chemical synthesis. However, enzymatic synthesis has some advantages in speed and use of non-hazardous ingredients.
DNA Script, Nuclera, Kern Systems, Ansa and Molecular Assemblies all use TdT technology. Other companies, such as SynHelix/Quantoom use DNA primase enzymes, while others employ proprietary geometric enzymatic synthesis instead (Camena Biosciences). Subsequent rounds of PCR and/or ligation can increase the size of genome fragments synthesized in a larger way. For example, Gigabases (from ETH Zurich) became the first entity to build a bacterial genome entirely designed by a computer algorithm. They did this through evolution guided multiplex DNA assembly (patent here). However, the cost of synthesis from Gigabases ranged from $30,000 for 100 kb to $400,000+ for genome level synthesis, a price far too high for many companies and labs.
After years of being a stalwart of the phosphoramidite platform, Twist has announced plans to develop its synthesis capabilities with enzymes. Although there is no commercially produced DNA from Twist synthesized with enzymes, the strategy expansion highlights the broader vision of DNA synthesis by Twist and may signal that enzymes are the future of DNA synthesis. Namely, Twist dovetails its goals in DNA storage with enzymatic DNA synthesis, invoking that the hazardous chemical used during phosphoramidite synthesis doesn’t align in a future with many decentralized DNA storage facilities.
Enzymatic synthesis is priced comparable to ink-jet printing and electrochemical synthesis, but has higher coupling efficiency. However, enzymatic synthesis is not without its foibles, one of which is parallelization. I haven’t been able to find any patents that parallelize enzymatic DNA synthesis, but there have been some publications which parallelize enzymatic DNA synthesis of 12 oligos simultaneously. There is almost certainly multiplexing in enzymatic synthesis but it’s unclear if it's been engineered to the magnitude of phosphoramidite synthesis. There is a case to be made where enzymatic synthesis doesn’t need to be parallelized as much due to the longer oligos.
And it doesn’t seem far away. Here’s a recent quote from the CEO and founder of DNA Script:
“DNA Script was the first company to enzymatically synthesize a 200mer oligo de novo with an average coupling efficiency that rivals the best organic chemical processes in use today. Our technology is now reliable enough for its first commercial applications, which we believe will deliver the promise of same-day results to researchers everywhere, with DNA synthesis that can be completed in just a few hours.”
While the continuous, long strands that enzymatic synthesis enables isn’t necessary as a standalone final product because short strands can be stitched together by companies like Twist, the enzymatic approach could enable benchtop DNA printers which empowers a far tighter feedback loop for lab coats. Indeed, researchers at Cal and Lawrence Berkeley Lab behind a novel enzymatic process say the ease of use could lead to ubiquitous “DNA printers” in research labs, akin to the 3D printers in many workshops today. “If you’re a mechanical engineer, it’s really nice to have a 3D printer in your shop that can print out a part overnight so you can test it the next morning. If you’re a researcher or bioengineer and you have an instrument that streamlines DNA synthesis, a ‘DNA printer,’ you can test your ideas faster and try out more new ideas.”
Here are other exciting advances in the industry:
- Twist has issued two patents which has the synthesis capacity of up to 1,000,000 nucleotides concurrently, two orders of magnitude higher than their current capacity, suggesting their aspirational technical goal. Other advancements include computationally predicting fragment parsing for the de novo synthesis of genome-scale DNA via pooled assemblies.
- GenScript launched the first commercial miniature semiconductor platform that uses integrated circuits to 8.4 million unique electrodes at over 2.5 million per square centimeter density. With four chips running in parallel, a staggering 6.7 billion base pairs of DNA can be synthesized in just two days
- Elegen is DNA assembly with microfluidics + solid-phase + enzymatic reactions that does 7kb and soon 20 kb; error rate (1:70,000); 7 day turn-around; here’s the patent
- Evolution-Guided Multiplexed Dna Assembly of Dna Parts, Pathways and Genomes was used to synthesize a bacterial genome at Gigabases (now RocketVax)
- Greenlight Biosciences focuses on high quantities of small segments of DNA and able to produce those at scale for vaccines and crop protection.
- Microsoft is advancing electrochemical DNA synthesis to achieve higher density of synthesis in a given area and thus greater throughput:
[3]Increasing synthesis density, that is, the number of synthesis spots on a fixed area, is the key to increasing the writing throughput and lowering its cost. The closer together these spots are on an array, the lower the synthesis cost of each DNA chain because the materials needed for the process can be used with more sequences.
The main challenge to increasing DNA writing throughput is to maintain control of individual spots without interfering with neighboring spots. Current DNA synthesis arrays are designed for generating a small number of high-quality DNA sequences with millions of exact copies and are achieved through three main array synthesis methods: photochemistry, fluid deposition, and electrochemistry.
In photochemical DNA synthesis, a photomask or micromirror creates patterns of light on an array, which removes the blocking group from the DNA strand. Liquid deposition, such as acoustic or inkjet printing methods, deliver the acid deblock to the individual spots. Both methods, however, are limited in the synthesis densities they can attain due to micromirror size, light scattering, or droplet stability. Electrochemical arrays, however, can leverage the semiconductor roadmap where 7 nanometer (nm) feature sizes are common.
In electrochemical DNA synthesis, each spot in the array contains an electrode. Once a voltage is applied, acid is generated at the anode (working electrode) to deblock the growing DNA chains, and an equivalent base is generated at the cathode (counter electrode). The main concern when scaling down the pitch between anodes is acid diffusion; the smaller the pitch, the easier it may be for acid to diffuse to neighboring electrodes and cause unintended deblocking at those locations. While commercial electrochemical arrays have demonstrated acid generation and minimal diffusion at micron-sized electrodes, it was not clear if this trend would continue indefinitely to smaller features.
To put theory into practice, we had chip arrays fabricated with the previously mentioned layout. These electrochemical arrays contained sets of four individually addressable electrodes. With them, we demonstrated the ability to control DNA synthesis at desired locations by performing experiments with two fluorescently labeled bases (green and red). If acid were diffusing unexpectedly, it would reach unintended spots and we would see one color bleed over other spots.
On an electrochemical array, we generated acid at one set of electrodes to deblock the DNA chain and then added a green-fluorescent base. In the next step, we generated acid at a different set of electrodes of the same array and coupled a red-fluorescent base to generate the image seen in Figure 3. As expected, we saw no bleed over, confirming we had no unintended acid diffusion.
On a separate array, we then demonstrated the array’s capability to write data by synthesizing four unique DNA strands, each 100 bases long, which encoded the motto “Empowering each person to store more!” Although the error rates were higher than commercial DNA synthesizers, we could still decode the message with no bit errors.
Our proof of concept paves a road toward generating massive numbers of unique DNA sequences in parallel. By injecting electrons at specific locations, we can control the molecular environment surrounding the electrodes and thus control the sequence of the DNA grown there.
All this technological innovation has yielded orders of magnitude cheaper DNA synthesis:
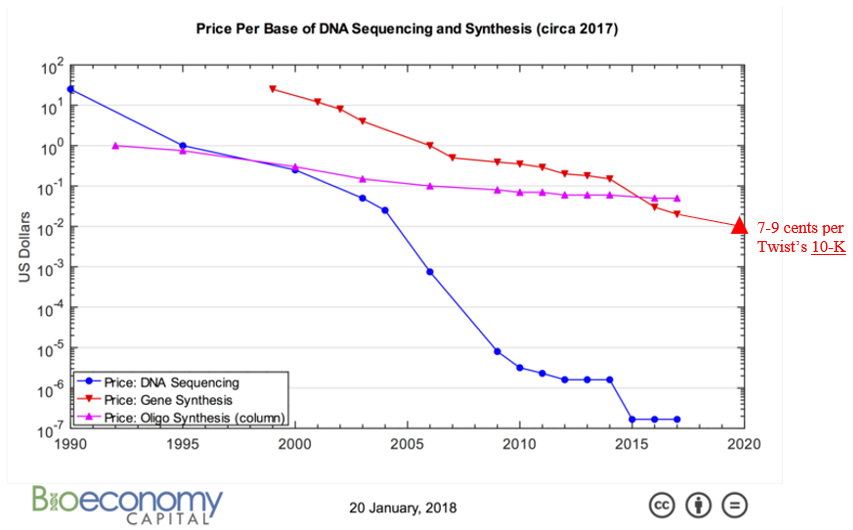
How quickly can we expect costs to continue coming down relies not just on continued innovation like those listed above but also on how much revenue the industry’s market dynamics generates to fund R&D and capex spend. Synthesis companies’ demand comes from orders for custom-made genes to develop biologic drugs, industrial enzymes or useful chemicals which are then produced in vats of microbes; researchers order synthetic DNA to insert into plants or animals or try out new CRISPR-based disease therapies. In other words, these companies grow alongside synthetic biology. However, whether their income statement grows in line with the broader ecosystem hinges primarily on whether or not the number of base pairs shipped outpaces their price decline. Years ago, an investor in the space penned a series of very thoughtful piece on the topic:1,1,1,1,1
What happens if the price of genes starts falling rapidly again? Or, forget rapidly, what about modestly? What if a new technology comes in and outcompetes standard phosphoramidite chemistry? The demand for synthetic DNA could accelerate and the total market size still might be stagnant, or even fall.
Which brings me to transistors. The market for DNA is very unlike the market for transistors, because the role of DNA in product development and manufacturing is very unlike the role of transistors. Analogies are tremendously useful in thinking about the future of technologies, but only to a point; the unwary may miss differences that are just as important as the similarities.
For example, the computer in your pocket fits there because it contains orders of magnitude more transistors than a desktop machine did fifteen years ago. Next year, you will want even more transistors in your pocket, or on your wrist, which will give you access to even greater computational power in the cloud. Those transistors are manufactured in facilities now costing billions of dollars apiece, a trend driven by our evidently insatiable demand for more and more computational power and bandwidth access embedded in every product that we buy. Here is the important bit: the total market value for transistors has grown for decades precisely because the total number of transistors shipped has climbed even faster than the cost per transistor has fallen.
In contrast, biological manufacturing requires only one copy of the correct DNA sequence to produce billions in value. That DNA may code for just one protein used as a pharmaceutical, or it may code for an entire enzymatic pathway that can produce any molecule now derived from a barrel of petroleum. Prototyping that pathway will require many experiments, and therefore many different versions of genes and genetic pathways. Yet once the final sequence is identified and embedded within a production organism, that sequence will be copied as the organism grows and reproduces, terminating the need for synthetic DNA in manufacturing any given product. The industrial scaling of gene synthesis is completely different than that of semiconductors.
DNA is always going to be a small cost of developing a product, and it isn't obvious making that small cost even cheaper helps your average corporate lab. In general, the R part of R&D only accounts for 1-10% of the cost of the final product. The vast majority of development costs are in polishing up the product into something customers will actually buy. If those costs are in the neighborhood of $50-100 million, the reducing the cost of synthetic DNA from $50,000 to $500 is nice, but the corporate scientist-customer is more worried about knocking a factor of two, or an order of magnitude, off the $50 million. This means that in order to make a big impact (and presumably to increase demand adequately) radically cheaper DNA must be coupled to innovations that reduce the rest of the product development costs. As suggested above, forward design of complex circuits is not going to be adequate innovation any time soon. The way out here may be high-throughput screening operations that enable testing many variant pathways simultaneously.
In summary, for the last forty years, the improved performance of each new generation of chip and electronic device has depended on those objects containing more transistors, and the demand for greater performance has driven an increase in the number of transistors per object. In contrast, the economic value of synthetic DNA is decoupled from the economic value of the object it codes for; in principle you only need one copy of DNA to produce many billions of objects and many billions of dollars in value.
The point above is important because for further price declines to happen, sufficient revenue must be generated by these companies to survive no less fund ever-larger manufacturing plants. The industry’s seen commendable growth since he wrote those pieces. GenScript now has 143,000 sq ft of synthesis facilities, 60% of which is fully automated, with total annual capacities of 2.4 billion bps vs the 1 billion he estimated the entire industry shipped in 2014. Aggregate synthesis market revenue rose from estimates of $50M in 2008 to $350M in 2014. Twist and GenScript alone will make $300M this year. All told, it’s a surprisingly small market with uncertain long-term economics. Its business model may need to change to capture more of the value being created, to intertwine the value of the final product with the number of base pairs used to make it. Otherwise, new manufacturing plants won’t break ground, industry R&D may slow, companies may go under, and the march downward in synthesis prices may slow.
Below is a spreadsheet with the top synthesis firms, the largest being GenScript, Twist, and IDT.
Note that DNA-based storage may be just the business model to turn the industry's economics around. The amount of data generated each year is already outstripping our ability to store it and that gap will only continue to widen. DNA is 1,000x denser data storage mechanism than the densest hardware humanity has invented – compact solid-state hard drives – and at least 300-fold more durable than the most stable magnetic tapes. As context, it could store a modern data center in test tubes or all the movies ever made in an area smaller than the size of a sugar cube. Additionally, whereas magnetic tapes must be replaced every 10 years, DNA lasts for tens of thousands of years and almost a million years when stored properly. See the addendum on DNA-based data storage for more info.
- Editing the Code of Life: CRISPR & Co.
As powerful as reading the code of life is, the ability to do so without the ability to efficiently edit it would be tantalizing, almost cruel. We’d have the power to understand the 75,000 genetic diseases, but hopeless to cure the hundreds of millions of people suffering from them; understand how to engineer more effective drugs, larger and more climate resistant crops, improved microbes for biomanufacturing, etc with the ability to act. We’d be Cassandra.
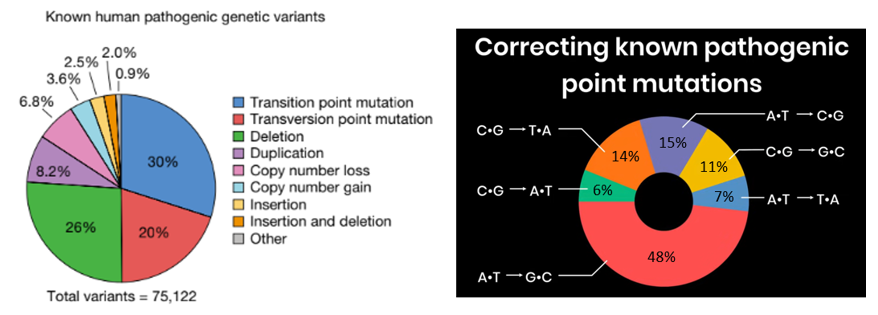
Thankfully, researchers have been developing mechanisms to edit genomes for decades with recombinant DNA taking off in the 70s, then TALENs, zin finger nucleases, and now CRISPR and her descendants. With each iteration, we can edit genomes more precisely with a broader range of edit lengths with fewer side effects on more cell types and species. Importantly, they also got exponentially easier to work with and entire industries were built around them such that high school biology students can now use CRISPR without breaking a sweat.
To give a sense for how much they’ve improved, consider that in gene editing’s humble beginnings, the efficiency rate of altering a gene by introducing a new molecule was about 1 in 1 million. Then with CRISPR-CAS 1, it got to about 1%. Techniques today achieves rates of 5-70%, depending on the technique, length of the edit, cell type, etc. For instance, one technique’s efficiency rate is 50-60% for cell lines and 4-5% in primary human hepatocytes and T cells. Regardless, the efficiency of gene editing has increased nearly a million-fold in its five-decade existence.
Note that the efficiency rates quoted above are only the rates at which it edits the DNA at all. When the system does make an edit, the rate of precision is the odds that it’s the intended one. This is far higher than efficiency rates and can exceed 99.9%.
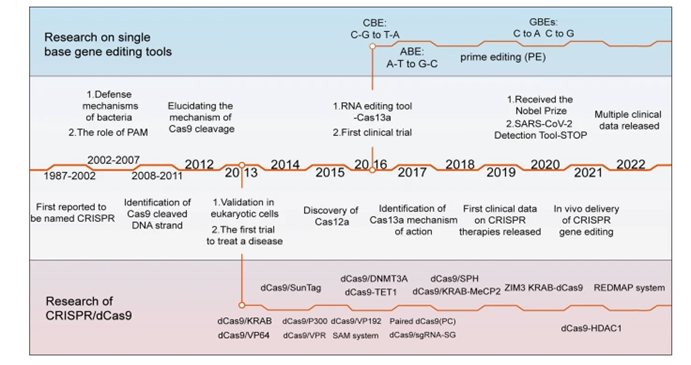
Though discovered in 2012, the origins of CRISPR dates back billions of years. Bacteria’s immune systems developed a mechanism to defend against viruses, with both a way identify the DNA as foreign and an effective scissor to cut it in two and cause it to dysfunction. It’s able to recognize various potential threat with pinpoint accuracy and to remember the threat for later in case it runs into it again. When a phage docks at a bacterial cell and injects its DNA into the cell, remnants of that code are inserted between repeating sequences of codes, a sort of library of past invaders.
Researchers gradually pieced the story above together over the last several decades. The adaptive immune system described was once thought to be exclusive to vertebrates.1 The realization that it had evolved billions of years before came when genome sequencing started to be applied to bacteria. Researchers in the 80s noticed an unusual motif containing short, repetitive DNA sequences in E. coli.1
Subsequent investigation found this was conserved across other prokaryotes1, and in 2005 the sequences between the repeats, termed clustered regularly interspaced short palindromic repeats (CRISPRs), were analyzed and found to be exact matches to phage genomes.1
In the early 2010s, researchers like Jennifer Doudna figured out how to reprogram the bacteria’s natural ⌘F mechanism to guide the scissors to a location on the genome of our choosing and won the Noble Prize in chemistry in 2020 for it. This molecular guide is a single RNA strand of 18-24 nucleotides that stops searching the genome when it finds the sequence that mirrors its own. Human-programmed CRISPR gene editing swaps that sequence out with our own. The ease with which this can be programmed is contrasted with previous gene editing tools like ZFNs and TALENs which both require the design and creation of a custom protein for every target DNA. With CRISPR, you just type out your desired sequence and hit ‘print’ on a synthesizer.
Once the CRISPR system has fixed onto the right sequence, the nuclease ‘scissor’ enzyme cuts the DNA on both sides of the twisting ladder. A broken helix triggers the cell’s repair mechanisms to try to fix it. Here’s how that process works:
The double-strand breaks (DSBs) are mostly resolved through one of the two major repair pathways: non-homologous end joining (NHEJ) and homology-directed repair (HDR)6,7,8. Although NHEJ can efficiently religate two DSB ends, it is error-prone and generates insertions or deletions (indels)9,10. By contrast, HDR precisely introduces desired alterations including insertions, deletions, or substitutions based on a DNA repair template11,12. However, HDR is restricted to the S/G2 phase of the cell cycle and is inefficient in most therapeutically relevant cell types11,13. Indeed, in most somatic cells, NHEJ outcompetes HDR in repairing the DSBs, resulting in a complex range of editing outcomes5,14.
If the desired genetic edit includes an insertion, those genes are delivered to the repair system in the hope that it’ll include them.
The simplicity and versatility of CRISPR–Cas systems has led to their rapid adoption as the most widely used genome-editing technology for site-specific DNA manipulations. Since its discovery, the system has been relentlessly improved upon, with countless variations being both discovered and invented1,2. To discover new systems, researchers comb1,1 through the metagenomic space of the thousands of known nucleases and are actively traveling the world to sequence more species. They can vary wildly across size, accuracy, off-target effects, versatility, etc. For instance, one nuclease can target 90% of the human genome, while the first version discovered covers less than 10%1. Our knowledge of what’s out there as well as our mapping of these systems’ evolutionary lineage helps us engineer better versions.
Researchers have applied advances in protein engineering, structure-guided mutations, direct evolution, and phage-assisted evolution.39,40 The field’s ceaseless optimization has been across both universal traits like efficiency and specificity and to develop systems for specific environments and purposes. A bulk of the work has gone into improving efficiency. Since they evolved in prokaryotes, wild-type Cas systems often do not perform well in the more complex environment of human cells. Examples of the latter include systems designed for transcriptional activation83, epigenome editing84,85, and changes to mitochondrial DNA87. Consider the following two excerpts, the first providing high-level examples of optimizing different features for specific purposes and the second digging deep into the progress made on one performance attribute essential to all missions – off-target effects:
Structure-guided engineering of the DNA-binding pocket of Cas12a and Cas12f increased indel frequency, resulting in more efficient human genome editors33,41,42. Notably, Cas12f has been engineered to generate a hypercompact class of Cas effectors (~1.4–1.6 kb) that are more amenable than other Cas proteins for in vivo delivery and expression33. Others have been tailored for missions like single-base editing (e.g. CBE and PE) or transcriptional regulatory tools (e.g. dCas9-effector).
The off-target activities of CRISPR–Cas9 depend on multiple factors, including the inherent specificity and amount of Cas9 protein [5], the length, content, and structure of gRNA[8,9], the targeted cell type and cellular state[10,11], and the target site[6]. In the past several years, a variety of approaches have been proposed to reduce off-target editing, together with the methods for detecting off-target effects. Readers can refer to recent reviews on off-target detection for an overview and comparison of these methods[12,13,14,15,16]. This review focuses on strategies and implementations reported to increase editing specificity. The strategies utilized for reducing off-target effects include Cas9 nuclease engineering[17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33], using natural Cas9 nucleases with high specificity[34,35], the utility of base editors[36,37,38] and prime editors[39,40], gRNA design optimization and/or modulation [8,9,41,42,43,44,45,46], control of Cas9′s activity via direct delivery (e.g., ribonucleoprotein (RNP)[20,47], virus-like particles[48,49,50,51], cell-penetrating peptide-mediated delivery[52], mRNA[53], and self-limiting circuits[54]), and combination with anti-CRISPR proteins or CRISPR inhibitors[55,56,57,58]. Among these strategies, direct or indirect engineering of Cas9 proteins represents the majority of efforts, evidenced by the relatively large number of studies toward this direction (Table 1). Below, we summarize and discuss recent advances in improving the specificity of CRISPR–Cas9-mediated gene editing through Cas9 protein engineering.
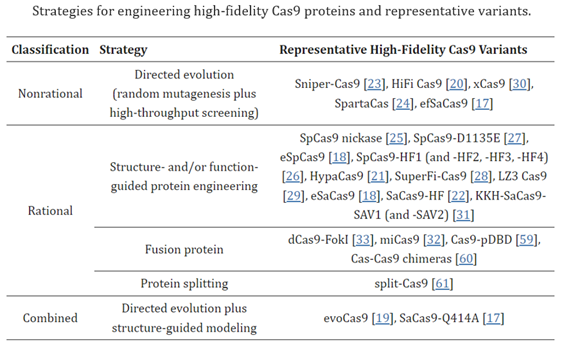
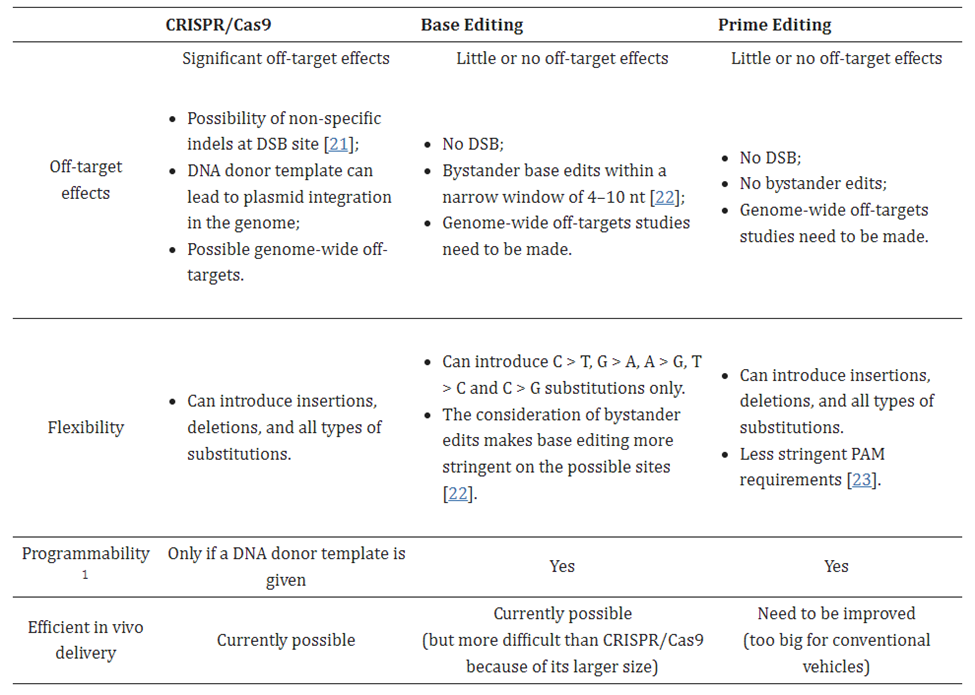
Despite the progress made on off-target effects, all CRISPR systems struggle in that regard compared to more recent gene editing systems, for reasons to be discussed. Their biggest class-wide con, however, is the fact that they make double-strand breaks (DSBs). While DSBs occur naturally in cells on several occasions1 and cells have mechanisms for dealing with them, they’re imperfect and can lead to unintended outcomes such as large deletions, inversions, and translocations, and the triggering of cellular stress response mechanisms to maintain genomic stability.1 To overcome this, some approaches rely on single-strand breaks generated by CRISPR–Cas9 nickase (nCas9, D10A or H840A) have been developed, albeit with low editing efficiency1. These inherent downsides of the CRISPR system highlight the need for more precise gene editing complexes with fewer unintended consequences.
In 2016, David Liu’s lab at Broad developed base editors (BE), a system that directly converts one base to another without inducing a DSB. It’s like a pencil eraser rather than scissors. No such mechanism exists in nature, so the researchers had to start from scratch. They took the programmable searching mechanism from CRISPR but deactivated its cutting capacity. A protein that performs a chemical reaction on the targeted base to rearrange its atomic structure was fused on. This directly converts one letter to another without disrupting the rest of the gene. At this point, only half the problem is solved as the other side of the double helix must be altered to reflect that change. The group further engineered the complex to nick the backbone of the opposite side of the double helix, tricking the cell into thinking the non-edited side was the one in error. The cell fixes that side and completes the transformation.
BEs have very high efficiencies and low off-target effects. They can execute single base substitutions for the four transitions (C>T; T>C; A>G; G>A), and recent studies have expanded this to include two transversions (C>G and G>C) 3,4,5. A major limitation is the inability to execute all 12 edits or to perform insertions or deletions.
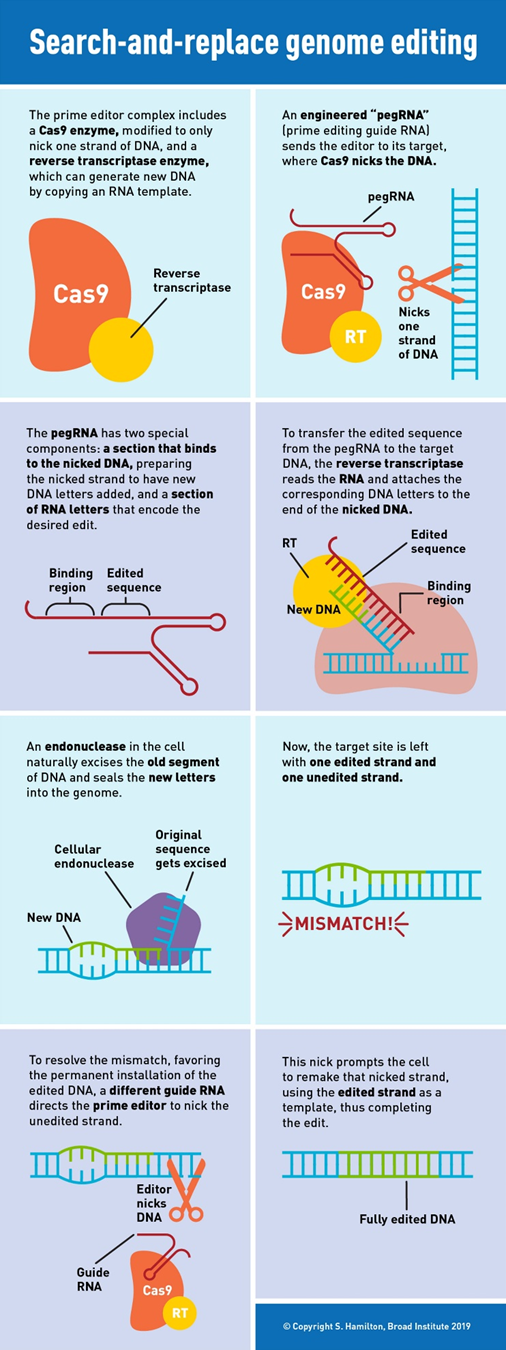
In 2019, Liu’s lab invented the next major class of gene editing technologies, prime editors (PE), by coupling a Cas9 to a protein called a transcriptase. A full breakdown can be seen to the right. PEs can make all 12 edits at single-nucleotide specificity, insertions, and deletions. This means they can in principal correct 94.4% of targetable pathogenic variants in the ClinVar database.1 Moreover, they rarely produce off-target errors or indel byproducts (typically <0.5% of editing outcomes), because unlike Cas nucleases and base editors, PEs requires three checkpoints of complementary base pairing for productive editing. As the CEO of Prime Medicine said, “it is unlikely that it would land on the wrong spot and unlock all three keys.”1 The limited off-target errors have been confirmed in mammalian cells (0-2.2%)60, mammalian organoids (0%)61,62, mouse embryos (<0.1%)63,64,65,66 and plants (0-0.23%)67.1 Indeed, preliminary results suggest that PE’s ratio of correct edits to unwanted byproducts may in the realm of 30x higher than that of Cas9.1
“With prime editing, we can now directly correct the sickle-cell anemia mutation back to the normal sequence and remove the four extra DNA bases that cause Tay-Sachs disease, without cutting DNA entirely or needing DNA templates,” said Liu. “The beauty of this system is that there are few restrictions on the edited sequence. Since the added nucleotides are specified by the pegRNA, they can be sequences that differ from the original strand by only one letter, that have additional or fewer letters, or that are various combinations of these changes.”
As of now, however, there’s no free lunch. Current PE systems have relatively weak efficiency. The first generation (PE1) had 0.7–5.5% base substitution efficiency. The second is typically below 20% in immortalized cell lines and drops even lower in primary cells. Note that 20% is a threshold generally considered important for therapeutic uses. Moreover, it varies wildly across target loci and cell types, slowing its broad adoption.
A second major drawback of the system is its hulking size. It’s too big to fit in the most commonly used and thoroughly studied delivery vehicle, an adeno-associated virus (AAV). Its guide RNA, called a pegRNA, is far larger than the sgRNAs used for CRISPR systems (>100 nucleotides vs 20). It must include both a primer binding sequence and the template for the desired RNA sequence. One group addressed the delivery problem by splitting PE into two parts and having split inteins proteins reassemble the parts into the complete complex. This approach was tested in mouse livers and retinas, with one group developing a compact PE.1,1,1 In a second approach, electroporation of a PE2 ribonucleoprotein complex (RNP) into zebrafish zygotes was shown to yield a 30% editing efficiency in somatic cells and even resulted in germline transmission.
Lastly, a double-edged sword of BEs and PEs, both of which don’t occur naturally unlike CRISPR-Cas9 and are the chimeric tethering’s of individually engineered molecules, is that they’re modular. On the one hand, specific pieces can be swapped out, boosting adaptability. On the other, that each component must be individually optimized.
All told, researchers have designed 35+ prime editor and pegRNA architectures. Most systems are optimal for 30bp long edits or less, which is convenient because 85–99% of insertions, deletions, indels, and duplications in ClinVar are of that length. Half a dozen different prime editors can precisely delete stretches of DNA up to 10kb or replace them with DNA inserts up to 250bp with efficiency rates of ~25%. One can insert up to 10kb sequences without any genomic deletion (only leaving attL and attR sequence scars from Bxb1 recombination during donor knock-in), though with lower efficiency. Other PEs can make alterations on the order of entire genes. One can invert 40kb of DNA between the IDS and IDS2 genes with 10% efficiency.
As two final points, we can also now edit other types of molecules. We can make precise edits in RNA up to thousands of bp’s in length, with some drug developers using this to correct the profile of proteins in cells. We also now have the ability to directly edit the DNA sequence of proteins after they’re constructed. And lastly, with all these methods, it can be confusing for researchers to determine which one to use for their specific needs. Wyss launched an integrated pipeline to help them figure out which genes to target, which tools to use, how to quickly clone and screen the targets, and how to interpret their results. The tool includes an algorithm that combines gene expression data with cell state and protein interactions, and assigns an elevated score to genes with strong connections to other genes and cell-level activity. A second algorithm generates networks to represent gene expression changes during cell differentiation and ranks key regulators using centrality metrics like PageRank
Read next section: DNA data storage
[1] Here's two other good technical overviews of the various synthesis methods: 1,2
[2] Here’s some more detail on how enzymatic synthesis works