Programming the Code Your Cells Execute: RNA & Gene Therapies (Century of Biology: Part II.I)
As said in Part I, DNA is like the master file to your body; changes to it are permanent and will affect all cells its host cell divides into. As such, it stays firmly put in the nucleus, safeguarded like the safe in a casino. Every cell in your body has that same master file. Only a small section of it is being executed at a given moment, and that determines whether it’s a lung cell or a neuron, whether it’s currently under Martial Law gearing up to go to war with an invading pathogen or humbly going about its day.
To make the world go ‘round outside of the nucleus, messenger RNA copies the section of the genome to be executed and shuffles into the bustling city that is the rest of the cell. There, it’s translated into proteins, molecular machines that do the actual dirty work that the DNA is calling for at that moment.
RNA and gene therapies harness these processes to control what code your cells are executing. They can both do so in four ways:
- Boost the expression of already present but insufficiently expressed genes
- Provide entirely new genetic material (for instance, the codes to construct the necessary antibodies to fight against the novel corona virus, an infectious disease your body’s immune system has never been exposed to)
- Silence dysfunctional genes
- Directly edit the genome or transcriptome
These new forms of therapy unlocks the entirety of the genome to therapeutics. Consider the following:
- 85% of the genome might be undruggable using small molecules1,1
- In the case of noncoding RNAs, especially small RNAs, few characteristics distinguish them from one another apart from their RNA sequence. RNA-based drugs, including antisense RNA and siRNA, operate via sequence-specific binding of their target molecules, suggesting that these drugs are likely to be the most effective when targeting noncoding RNAs. Because there are more noncoding RNAs than proteins in the human genome30, and because recent studies have confirmed the essential roles of these noncoding RNAs in the pathogenesis of various human diseases31,32,33, the importance of RNA drugs that can target these molecules will only increase.
- Fewer than 10% of rare diseases have FDA-approved treatments. About 80% of rare diseases are caused by known alterations in a single gene (NIH)
- “Of more than 50,000 genetic changes currently known to be associated with disease in humans, 32,000 of those are caused by the simple swap of one base pair for another.” – David Liu
- By targeting RNA, currently undruggable cancer drug targets like MYC and KRAS—which are overexpressed in 75% of cancers—can be revisited and potentially blocked before the protein is ever even produced. This example isn’t cherry picked. Currently, small molecules and antibodies only target 0.05% of the human genome—meaning that most diseases don’t have defined drug-binding sites. (Century of Bio)
With the ability to write, delete, or edit genetic outcomes, these therapies can each in principal address effectively any disease. Doctors could take a sample of a cancer patient’s tumor and then deliver a sequence encoding for the production of antibodies that direct their immune system to attack that malignancy. New pathogens, like COVID, could quickly have their DNA sequenced and a vaccine to defend against it produced. Genetic diseases ranging single base pair mutations to many kilobases could be cured at their source. Though these possibilities aren’t all realities yet, they’re now within reach thanks to the five decades of work across critical issues like delivery systems, safety, and expression levels and duration. Indeed, thousands of therapies are being developed to treat diseases across liquid and solid cancers, infectious diseases from COVID to the flu to HIV, immunotherapies, protein deficiencies and dysfunctions, inherited and acquired genetic diseases from cystic fibrosis to an inherited blindness, Parkison’s, Alzheimer’s, rare diseases, and more. You can solve a lot of problems if you just deliver the right code. See these reviews1,2,3 for details on the latest in RNA therapy applications and these for gene therapies.
This new therapeutic paradigm unlocked by delivering your cells the instructions they need as a payload and allowing the body’s natural factory-like production capabilities to do the rest can be juxtaposed to traditional therapeutics. Small molecules, proteins, or antibodies deliver the final outcome as a therapy. A patient missing a protein like insulin has that protein directly injected into their body; a patient with a dysfunctional protein swallows a pill that binds to and silences that protein. In comparison, RNA and gene therapies deliver the instructions encoding those actions as the payload. They’re thus like giving the cell a fishing pole instead of a fish. They’re abstractions further upstream from the ultimate desired outcome. And, gene therapies, by delivering DNA to sit in the nucleus and by transcribed into mRNA, is upstream of RNA therapies. Note that the further upstream you go, the longer lasting the therapy is. A pill delivers a one-time boost, mRNA can pump out proteins for up to a couple months, genes delivered into the nucleus can last years, and direct edits to the genome itself can be permanent. These approaches mean that the amount of the desired molecule produced isn’t directly proportional to the size of the pill or serum injected. For however long those instructions last, the cell will continue to churn out the desired output.
While both classes of therapies can perform the four edits mentioned above, their core difference is their duration of expression, which can be a good or a bad thing depending on the application. More transient therapies are safer but must be administered more frequently if the desired outcome is to be expressed for more than a couple months. On the other hand, therapies that venture inside the nucleus run the risk of messing with the genome in deleterious ways, from inserting next to and promoting the expression of mutated cancer encoding genes to insertional mutagenesis to on- and off-target errors. However, direct edits to the genome, if made correctly, are true cures to genetic diseases. A sickle cell anemia patients may never have to think about it again once a base editor makes that swap.
The following sections will explore the therapies from most transient, short-lived RNA therapies, to most permanent, direct genome editing.
RNA Therapies
A cell’s RNA profile is like the code it’s actively executing. These therapies have the benefit of being safer than gene therapies due to their transience and because they don’t rely on viruses to be delivered. They’re also much cheaper because they’re manufactured in a cell-free way. They can even be cheaper than traditional therapies like antibodies. Having cell do the manufacturing can be much cheaper than doing it in a lab. For instance, the cost of producing enough antibodies for billions of doses of COVID vaccines would have surely been far more expensive than having the body do it for us. RNA therapies may also unlock new business models thanks to the simplicity of the core payload delivered: a string of letters. The tricky part is delivering them safely and efficiently to the right cell. Once its chemical structure and mode of delivery are standardized, it may prove relatively easy to write the next sequence. This may make it economically sustainable for therapeutics companies to go after very rare diseases.
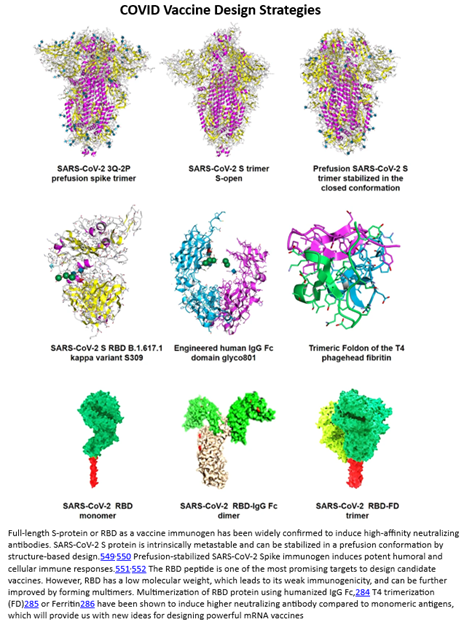
These points were largely vindicated by the dramatic success of the mRNA-based COVID vaccine. From the moment researchers got hold of the virus, its DNA was sequenced within days, initial designs for a vaccine were produced within weeks, finalized vaccines safe and effective enough to be tested in humans were produced in months, and in under a year a product with 90%+ efficacy and tremendous safety characteristics was rolled out globally. To elucidate exactly how such a vaccine is different from traditional ones, consider that the COVID vaccine delivers the genetic sequence encoding the production of antibodies to target the viral spike protein unique to COVID as seen to the right. Whereas, traditional ones would inject small quantities of the whole virus, either in the form of inactivated viruses, attenuated live viruses, or genetically engineered viruses, into the host body. To date, nearly ten billion doses have been manufactured and sold at the price of a Chipotle bowl. And, as vindication of the concept that once the delivery mechanism is solved for the payload is highly adaptable, vaccines to new variants were rolled out in fractions of the time of the original one.
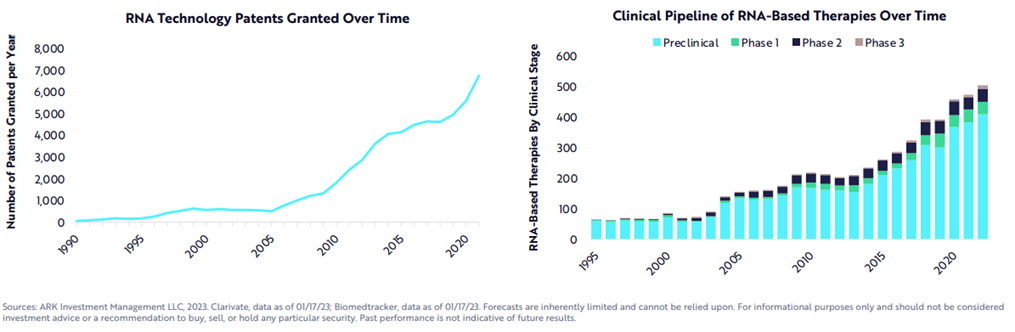
While the COVID vaccine made RNA therapies world famous, a range of modalities exists to treat the four classes of edits, as seen in the table below.[1] In fact, the first RNA-based drug was approved over 20 years ago. Since then, over a dozen products have been approved with nearly all in the last six years, a thousand companies launched with thousands of assets in the pipeline supported by tens of thousands of patents and nearly $100 billion in funding raised.
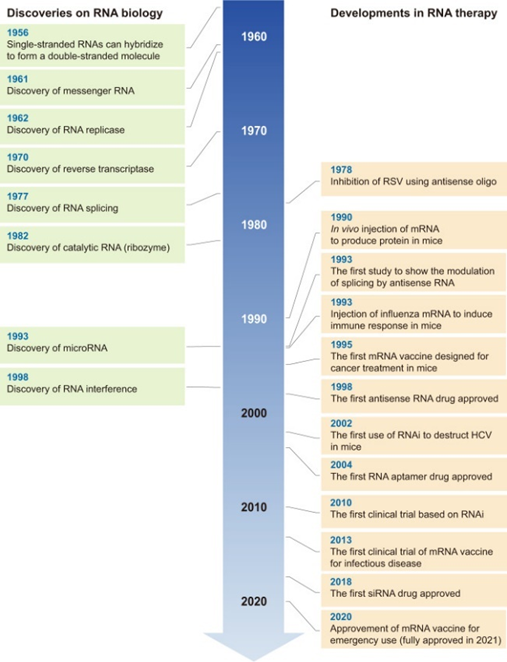
All this work has recent success is built on five decades of progress in science and engineering to improve the RNA’s stability, their delivery systems, their safety and efficiency, manufacturing, and more. The key breakthroughs have been with respect to chemical structure modification to radically improve RNA durability (nucleases degrades wild type RNA within hours), sequence optimization12,13, purification14, cell-free manufacturing, and lipid-nanoparticle (LNP) delivery design. Here’s some more detail:
Chemical modification is one of the most promising strategies for delivering RNA-based drugs. Modification of the nucleic acid backbone, ribose ring, and nucleobase itself has been widely used to optimize drug-like characteristics for enhanced delivery. For example, extensive chemical modifications allow the delivery of gapmer ASOs to various tissues without an extra delivery agent. To date, eight of the ten approved oligonucleotide treatments are applied without an additional delivery vehicle. However, some unnatural nucleotides may be harmful. For instance, LNA-modified nucleic acids were found to cause severe hepatotoxicity [164]. Therefore, bioengineered RNAi agents (BERAs), a newly-developed in vivo RNA agent carrying no or minimal post-transcriptional modifications, have shown good application prospects [165]. (LINK)
NLPs notoriously get soaked up by the liver, which evolved to filter out these types of particles. With current technology, less than 5% of the particles reaching the targeted disease tissue. While developing new approaches for liver detargeting is an active area of research, this shortcoming has skewed early gene therapy trials towards liver diseases—much like how the delivery problem led to a greater focus on retinal disease.
Plenty of challenges of course remain, as reviewed here:
Therapeutic mRNA is synthesized via in vitro transcription (IVT) in a cell-free system using a linear DNA template generated from a plasmid or PCR and RNA polymerase T7, T3 or SP6 (ref. 30) (Fig. 1a). mRNA is then purified using conventional laboratory-scale purification methods for nucleic acids (for example, precipitation in ethanol). However, these methods often fail to remove impurities such as double-stranded RNA and RNA fragments, which reduce the therapeutic efficacy and cause undesired biological responses in clinical use31. Upon local or systemic administration, mRNA can be rapidly degraded by the abundant nucleases in the extracellular space, removed by macrophage phagocytosis or cleared by renal filtration24,32,33 (Fig. 1b). In the meantime, mRNA is a large, very negatively charged, single-stranded polynucleotide that is difficult to pass through negatively charged cell membranes. In fact, only 0.01% of extravasated mRNAs from blood vessels can enter target cells2, where most of the mRNAs are trapped in endosomes and degraded thereafter (Fig. 1c). Eventually, a fraction of the internalized mRNAs escape from endosomes and reach ribosomes for therapeutic protein translation.
The immunostimulatory potential of exogenous mRNA is another major hurdle to clinical translation15,18 (Fig. 1c). Exogenous mRNA can be sensed by pattern recognition receptors (PRRs) that play a crucial role in responding to viral RNAs, inducing immune stimulation34. Upon endocytosis, mRNAs can be detected by endosomal sensors called Toll-like receptors (TLRs)35, the main family of PRRs expressed primarily but not exclusively by immune cells. mRNAs that escape endosomes can be sensed by several cytosolic PRRs. Stimulation of these PRRs eventually results in the production of type I interferons (IFNs) and other proinflammatory cytokines36. These secreted IFNs bind to their receptors on the stimulated cell and adjacent cells—activating the JAK–STAT pathway, which triggers the transcription of over 300 IFN-stimulated genes. Among these, IFN-inducible protein kinase RNA (PKR) can suppress activity of the translation initiator initiation factor 2, leading to inhibition of mRNA translation37,38, and the 2′-5′-oligoadenylate synthetases39 and RNA-specific adenosine deaminase40 can reduce the stability of mRNAs. Because all of these challenges have substantially limited the clinical use of mRNA, advances in mRNA-related technologies are needed to address these challenges before the full therapeutic potential of mRNAs can be unleashed.
And, here are some of the exciting recent innovations in mRNA design:
Self-amplifying mRNA contains an alphavirus-based replicon that can amplify the expression of encoded proteins, and therefore requires a much lower dosage than conventional mRNAs in most applications110,111. The incorporation of additional replicon genes makes the size of self-amplifying mRNAs larger than conventional mRNAs. Thus, the formulations used for conventional mRNAs may need to be further optimized for the larger-size self-amplifying mRNAs111,112. While nucleoside modification is widely used in currently approved or currently investigated mRNA-based therapies, self-amplifying mRNAs cannot contain these modifications because they can interfere with the self-amplification process113. Self-amplifying mRNA-based SARS-CoV-2 vaccines have already demonstrated their ability to induce high neutralizing antibody titers in animals114 and several candidates are currently being tested in clinical trials48. These have the potential to be used at lower doses (1–10 µg) than conventional mRNAs (30–100 µg) in COVID-19 vaccines113.
The unique closed ring structure enables higher stability of circRNAs compared with linear mRNAs, due to the lack of end motifs required for exonuclease-mediated degradation120. Indeed, a pioneering study showed that a circRNA constructed using a self-splicing intron exhibited robust and stable protein expression in eukaryotic cells121. The self-circularization of the linear RNA precursor eventually results in circRNAs containing an internal ribosomal entry site (IRES) to drive the expression of proteins121,122 (Fig. 3a). Besides the enhanced stability, circRNAs induce far fewer undesirable immune responses than unmodified linear mRNAs since they do not activate RNA sensors such as TLRs and retinoic acid-inducible gene-I123. A circRNA vaccine has elicited a higher level of neutralizing antibodies than a linear mRNA vaccine, demonstrating great protective efficacy against SARS-CoV-2 and its emerging variants in mice and rhesus macaques124. More recently, circRNAs containing five key elements, including vector topology, 5′ and 3′ UTRs, IRESs and synthetic aptamers, were constructed125, and simultaneous optimization of these elements resulted in higher circRNA protein yields125. In addition, several companies are exploring other variations on circRNAs, such as optimized IRESs (Fig. 3b).
That same review then covers innovations in mRNA delivery, including an endogenously produced protein carrier, improved LNPs, biological membrane-based vehicles like exosomes, organ- or cell-specific delivery, and inhalable, intranasal, or oral delivery systems.
- Gene Therapies
The field has been actively pursued since the 80s but has only become feasible to systematically roll out in the last 5-10 years thanks in large part to advancements in delivery systems. These systems must safely transport the DNA strand through the body to the right tissue and even the right cell type all without triggering the always-vigilant immune system, designed specifically to detect and eliminate foreign molecules. Once at the right cell, they must get through the cell membrane[2] and execute its job. If that’s altering the cell’s genetic code, it must also navigate into the nucleus, find the right DNA section, and make the edit without any off-target errors. Additionally, the transferred gene must be expressed at the appropriate level (neither too low nor too high) and for the appropriate duration to mediate the desired clinical effect.
Given the task’s complexity, we’ve designed our delivery systems based off the solution to delivering genetic payloads to cells nature optimized over millions of years: viruses. Researchers keep the outer cartridge that envelope the DNA and transports it to the right cell while removing the virus’ disease-causing genes and replacing them with the gene(s) being transferred and the sequences that control its expression. Making these systems safe and effective has required many decades of carefully studying how they work and engineering them to express our desired traits. Viruses are one of the threats that our immune system is the most primed to destroy, limiting their efficacy and posing serious toxicity risks. As an example of how far these systems needed to come, one of the more commonly used viruses today is based on the HIV virus, which has killed about 32 million people to date. It must be engineered to remove as much of its toxicity as possible while retaining the useful parts.
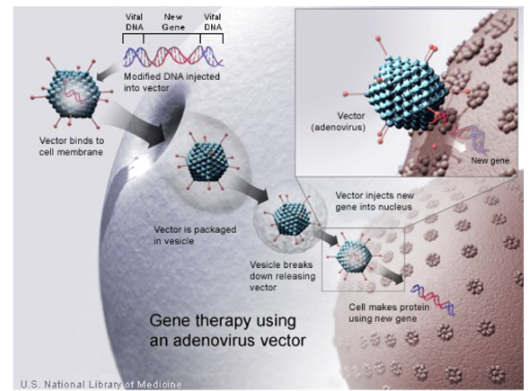
Nearly all gene therapies are based on one of three vector types: adeno-associated-virus (AAV), adenovirus (Ad), or lentivirus. These viruses were first discovered in the 50s and 60s. From there, basic biological investigation began slowly; in ’72, researchers first proposed gene therapies1; the 80s saw the first cloning and sequencing; by 90s they were ready to be tested in clinical trials in humans; then more systematic investigation ramped up with advent of HTS, intentionally scanning the animal kingdom for other variants, etc. These systems are engineered to avoid and circumvent the following limitations: strong immune system responses, pre-existing immunity, tissue / cell specificity, cell entry efficiency, gene transfer efficiency & longevity where applicable, payload size constraints, the inability to re-dose, complications involved in scaling manufacturing.
Engineering efforts start with the genome, iteratively removing all unnecessary and dangerous genes, allowing for safer vectors with higher genetic payloads. For instance, look at the engineering of Ad over time, as seen to the right. Over three generations of vectors, researchers progressively figured out which genes could be removed without making the delivery mechanisms impotent. It’s resulted in one version in which effectively the entire viral genome has been deleted, with much of that functionality being transferred to a secondary helper vector. This allows for a huge payload cargo when that’s of importance.
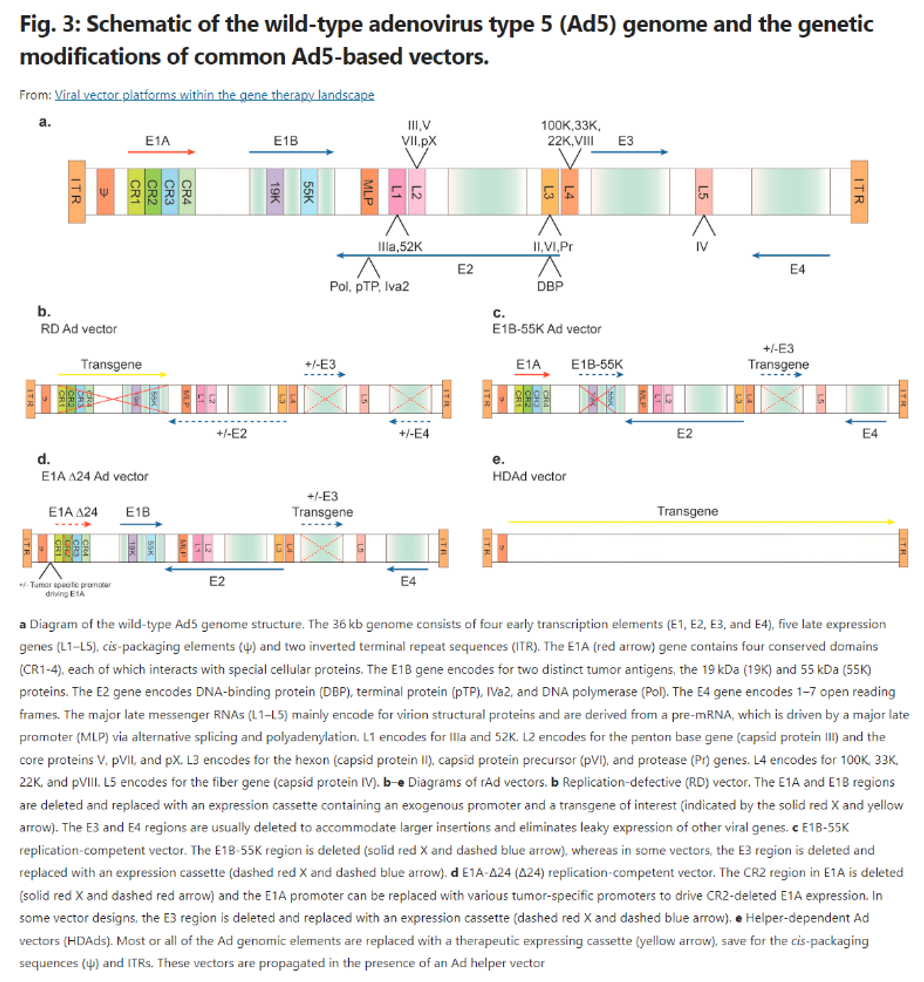
Other engineering efforts relate to making the protective shell better at navigating to the correct tissue / cell without triggering an immune response. For instance, consider the work done to develop improved AAV capsids1:
i. Natural isolates: Serotypes AAV1 and AAV2 were first discovered from tissue culture. Afterwards, AAV serotypes were isolated from a range of mammalian species and tissue types. Between 1965 to 2001, only a handful of AAVs, mostly from human clinical samples, were derived from viral isolates, vectorized, and tested. Following these efforts, which demonstrated that each serotype had unique tropism profiles, larger studies to isolate AAVs from human and non-human primate tissues were accomplished by using PCR- amplification of proviral cap sequences. At the end of 2009, more than a hundred variants across six different clades were discovered.131,175,176 Although only a few research groups continue to screen natural variants as potential gene therapy vectors, they are still the predominant capsids used in clinical studies today.
ii. Rational design: The engineering of pre-existing capsids by rational design is an efficient means of developing new capsids with specific properties.177 Such strategies began with the grafting of peptide sequences known to bind surfaces of target tissues onto the surface of the capsid.178 The latest achievement using such a strategy was the development of bone-homing capsid named AAV9.DSS-Nter.179 However, the generation of functional capsids through these means are in general difficult. Part of the reason behind this difficulty is that the AAV capsid and it’s role during the viral life cycle is not completely understood, i.e., changes on the capsid may theoretically improve binding of AAV to specific cell receptors, but may negatively impact other aspects, such as vector production or cellular trafficking.
iii. Directed evolution: Iterative selection of randomly mutated capsids in cell culture or in animals, coupled with next-gen sequencing strategies have given researchers a manageable means of developing new capsids with defined properties.177 Randomized mutations can be achieved by error-prone PCR180 or by the generation of chimeric capsids (capsid shuffling) from the primary sequences of pre-existing serotypes.181,182,183 The most noteworthy examples of evolved capsids are derived from platforms based on the insertion of short, randomized peptide sequences onto the threefold protrusions, which are highly amenable for capsid manipulation.184 Among the first in this class of capsids are AAV2.7m8 and AAVPHP.B.185,186 The AAV2.7m8 capsid was derived by the insertion of a random seven-amino acid sequence into loop VIII of the AAV2 capsid and iterative rounds of random mutagenesis and selection of positively transduced photoreceptor cells.
The development of high-throughput sequencing approaches in the early 2000s marked not only the capacity to profile the transcriptome and epigenetic marks on a whole-genome scale but also allowed for AAV investigators to expand screening depth to test millions of variants at once. Unfortunately, these screening methodologies are limited in that they can only evolve capsids that perform well in the systems they are screened in. For example, the discovery of AAV.PHP.B, which was screened for its ability to traverse the blood–brain barrier (BBB) in C57BL6 mice, produced a capsid that exhibited strain-specific differences.187 Nonetheless, this work was able to reveal the species- and strain-dependent LYS6A gene as the critical receptor that permits AAV.PHP.B to traverse the BBB.188 Additional capsids have thus far been evolved that do not rely on Lys6A host expression, such as AAV-F.189 Further improvements to these methodologies are ongoing and they offer a rapid means of developing new capsids for specific targets.
iv. In silico approaches: The use of machine learning approaches truly represents a new frontier in the gene therapy field and has great potential for producing capsids sequences not seen in nature. These means can potentially bypass the iterative steps required for directed evolution and can overcome the trial and error process attributed to rational design approaches. Machine-guided strategies have already demonstrated their utility by uncovering a previously unknown ORF encoding for the MAAP gene128 and by the discovery of Anc80, a capsid whose sequence is predicted to be the “ancestral” capsid by phylogenetic connectivity between contemporary capsids. Importantly, Anc80 has shown strong tropism to the mouse liver, muscle, retina, and cochlea.190,191
Choice of viral vector depends on the size of the gene or genes that it can carry, the target cells (dividing or nondividing, and cell type), whether the virus will insert into the target cells’ genome or remain separate, and the antibody status of potential patients. In addition to viral vectors, some gene therapies use non-viral vectors like LNPs and exosomes.
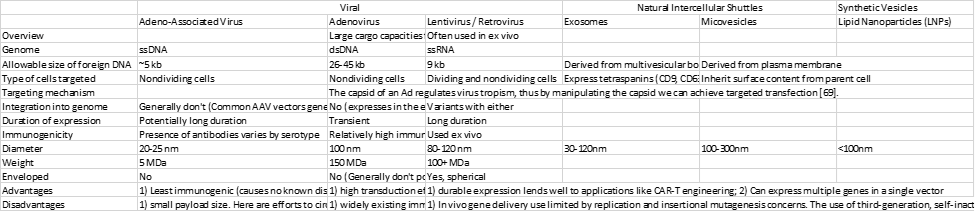
A final major challenge for gene therapies across vectors is their extremely high costs thus far, generally in the $100,000s, but can reach $2M. The therapies’ manufacturing complexity (in part due to its reliance on viral vectors), the fact that it has only just recently left the lab for commercial-scale rollouts, and that it has targeted rare diseases has driven its prices. Today, a typical manufacturing run of an AAV-vector therapy using high-yield cell lines and large-capacity bioreactors might only produce approximately ten doses of a systemic gene therapy from a single batch at a cost of nearly $100,000 per dose (assuming approximately 1 × 10^17 vg per batch).12 The industry has improved its efficiency by, for instance, lowering the frequency of empty capsids, which have no cargo and thus create the need for higher doses (increasing costs and immune response). Such processes include machines to automatically measure molecular weight and charge and sort out the empty ones, and the engineering of cell lines that package full capsids more efficiently. To see virus-specific manufacturing challenges, look at the Disadvantages section of the table above. Non-viral systems generally have the benefit of cheaper production. Gene therapies’ costs will gradually decline as they gain in scale, expand beyond rare diseases, find alternatives to viral vectors when appropriate, increase manufacturing process efficiency, and delivery efficiency (which will drive down dosing).
Equally as exciting as the improvement in delivery systems, the advent of CRISPR, BE, PE and the like brings fundamentally new opportunities to edit the genome in increasingly safe and precise ways. Previously, gene therapies altering the genome relied on viral vectors that naturally integrated into the genome, like retrovirus. However, these viruses couldn’t be programmed with specificity like the gene editing protein complexes. They generally integrate in places along the genome that the viruses evolved to target, each with its own preferences.1,2,3,4 Unable to control the exact integration location, these viruses sometimes integrate next to genes that promote tumor growth or interrupt proper transcriptional networks. Hence, genome editing using viruses has been largely limited to ex vivo applications, where patients’ cells are edited outside their body and can thus be screened for proper integration before putting them back in.
In comparison, modern gene editors can be programmed with single-base pair specificity. Groups have been developing gene-editing therapies across CRISPR, BE, PE, ZNF since 20091. The first ex vivo CRISPR-based clinical trials began in 20171, in vivo trials began in 20201, and the first BE-based trials began last year1. The tricky part with in vivo editing efforts has been delivery1,2, as these systems are too big to fit in an AAV.[3] Groups have addressed this in various ways, including splitting the sgRNA and the Cas protein into two different AAVs, delivering mRNA encoding for one or both in a LNP, and engineering the nuclease to be as small as possible.1 Combining the advancements in editors and delivery mechanisms has resulted in progress towards true cures for genetic disease[4]:
Sickle cell
Exa-cel is a potential one-time treatment for sickle cell disease and β-Thalassemia, two blood-based disorders that distort the production and structure of hemoglobin, the protein in red blood cells that carries oxygen. Without enough normal hemoglobin, red blood cells change shape, allowing them to clump together and block blood flow. Patients often require frequent transfusions to restore blood volume levels and oxygen flow. Exa-cel edits the patient’s stem cells to produce higher levels of hemoglobin, helping to alleviate or eliminate transfusion requirements for patients.
In its latest update, CRISPR and Vertex reported that 42 out of 44 participating patients with β-Thalassemia are transfusion free after treatment, with follow-up ranging from 1.2 to 37.2 months. This result represents 95% efficacy, higher than the 89% for bluebird bio’s Zynteglo, a one-time β-Thalassemia gene therapy approved by the FDA in August 2022.22
In sickle cell, all 31 patients treated with exa-cel were free of vaso-occlusive crises, with follow-up ranging from 2.0 to 32.3 months. On average, trial patients had hemoglobin levels of 43% 12 months after infusion with strong safety data.23 (Source)
Permanently lowering blood cholesterol
Cardiologist and Verve co-founder Sekar KathiresanMD, highlights three insights into cardiovascular disease that have emerged from human genetics research. First, if an individual’s blood cholesterol level is low throughout their lifetime, they are very unlikely to develop a heart attack. Second, some individuals resistant to heart disease carry mutations—resistance mutations—that naturally turn off cholesterol-raising genes. Third, three of the resistance mutations affect the low-density lipoprotein (LDL) cholesterol pathway, four of them affect the triglyceride-rich lipoprotein (TRL) cholesterol pathway, and one of them affects the lipoprotein(a) (Lp(a)) cholesterol pathway.
“CRISPR technology allowed us to develop a medicine that would mimic these natural resistance mutations. Our once-and-done gene editing medicines are designed to permanently lower blood cholesterol.”
VERVE-101, the company’s most advanced product candidate, targets the PCSK9 gene to lower LDL cholesterol levels. VERVE-201, the company’s next most advanced product candidate, targets the ANGPTL3 gene to lower TRL cholesterol levels. Both the VERVE-101 and VERVE-201 programs rely on base editing technology. To target the LPA gene and lower Lp(a) cholesterol levels, Verve is designing a custom editor.
VERVE-101 is currently in a Phase I trial, called heart-1, to evaluate safety and efficacy in patients with heterozygous familial hypercholesterolemia (HeFH), a genetic form of high cholesterol and premature heart disease. A morbid disease, HeFH is characterized by severely elevated blood levels of LDL cholesterol and premature atherosclerotic cardiovascular disease. Current LDL cholesterol–lowering regimens of daily pills or intermittent injections place a heavy treatment burden on patients, providers, and the healthcare system, and fewer than 5% of HeFH patients globally attain LDL cholesterol goal levels. (Source)
In addition to directly editing the genome to modulate gene expression, some groups have began targeting epigenetic networks with the aim of being able to turn gene expression off and on. ____. CRISPRa has been shown to increase regulation efficiency in vivo by as much as 5x and CRISPRi downregulated by 3x.1
Compared to other existing strategies for the therapy of genetic or acquired diseases, CRISPRa-based approaches offer several important advantages: (i) They are independent of target gene size and can be used in multiplexing experiments to activate different genes simultaneously. (ii) They induce little to no off-target effects in vivo18,19,20. This is partly because potential off-target sequences, in addition to matching the target sequence, must also be part of a promoter or enhancer region. (iii) They can be used for simultaneous activation and knockdown of different genes in target cells. This requires the catalytically active Cas enzyme and the use of sgRNAs with spacer sequences of different lengths. For spacers less than 16 bp in length, Cas loses its catalytic activity, but the CRISPRa module is still capable of gene activation6,37. Such an approach can be used, for example, to treat gain-of-function mutations that require simultaneous knockout of the mutant allele in addition to gene supplementation. (iv) In contrast to CRISPR/Cas modules which rely on the endonuclease activity, CRISPRa generally do not induce permanent changes in the genome. While permanent modification of the target locus is the desired effect in some diseases (e.g., to accomplish efficient gene knockdown), this can also become a problem if such modifications cause undesirable effects (e.g., large deletions) or occur at undesirable locations in the genome38,39,40,41,42.
As context for where this technology may go, consider the response to the question where CRISPR gene editing will be in ten years from the executive director of the Jennifer Doudna-founded Innovative Genomics Institute:
I think we will see much more epigenetic editing, using CRISPRa to activate genes and CRISPRoff to turn off genes. I believe that this will be the key that unlocks cures to common and complex diseases such as cancer and neurodegenerative diseases. Wouldn’t it be great if we could go into an individual’s genome and systemically control gene expression, rather than clipping genes and permanently turning off their expression? We may not fully realize this in 10 years, but I think we will be in a very different place with epigenetic editing in five to 10 years than where we are today.
Finally, remember that for even more impermanent gene editing without any risk of genomic mutations or mutagenesis, one can modulate the transcriptome via CRISPR-based mRNA editing.
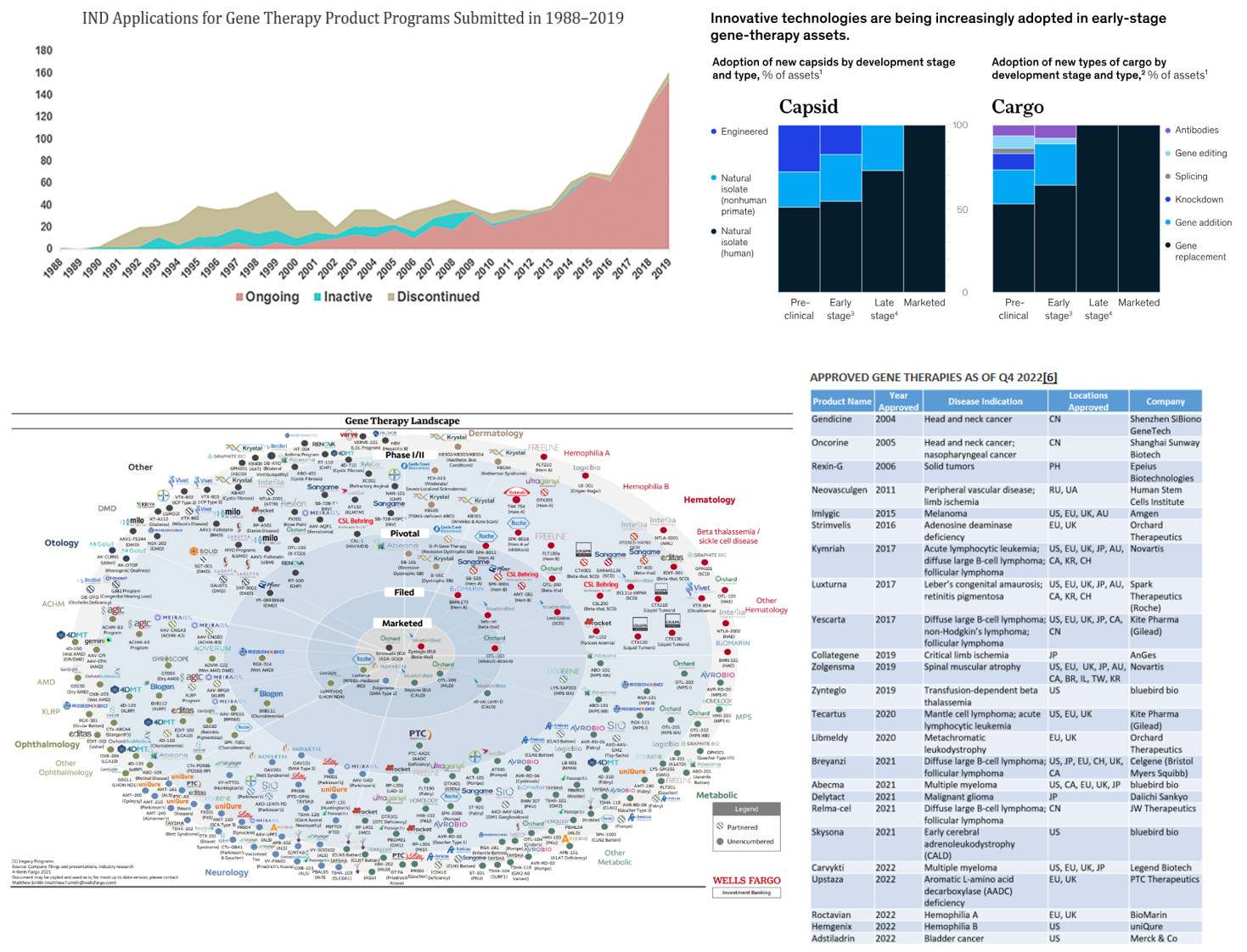
All told, half a century after the work towards gene therapies began and three decades after the first clinical studies, the drug class has finally advanced to the point of being systematically rolled out. The FDA has approved 13 therapies, five of those in the last two years. Moreover, the FDA has repeatedly expressed its bullishness on the drug class and its intent on helping safe and effective drugs pass through trials quickly. For instance, the head of the FDA’s Center for Biologics Evaluations and Research said that if the FDA only approves two or three per year going forward, "that’s a failure. Success would be if not exponential, at least some logarithmic progression towards more and more gene therapies being approved.” To get there, the CBER has a four-point plant: improve manufacturing through new research, clarify the use of the accelerated approval pathway for gene therapies, coordinate regulatory decisions with other regulators and launch an Operation Warp Speed for rare diseases. To date, FDA has approved over 300 accelerated approval applications.
Read next section: cell therapies
[1] For more detail on each type, see these reviews1
[2] Molecules enter the cell through endocytosis and fusion. Cells have evolved specific machinery to ingest materials from their environment and release some of their inner contents. The ingestion process is called endocytosis. Some cells constantly eat things in their environment—macrophages eat 25% of their own volume of fluid every hour. While this rate is extreme, all cells are continually taking small sips of their environment by pulling in small portions of their own membrane.
Despite this ingestion, cells maintain a constant volume and membrane size because they continually release materials via exocytosis. Cells internally form membrane-bound payloads that are transported to the cell membrane, where their cargo is either incorporated into the cell surface or secreted into the environment.
Certain nanoplexes and cubosomes have this inherent fusogenic property that enables better cell penetration[2], [35], [36], [37].
[3] ZFNs and TALENs don’t require dual AAV vectors for in vivo expression due to the small size of these proteins1, however, they require new protein designs for each target. Another key advantage of CRISPR-Cas-based systems over ZFNs or TALENs is the ease of multiplexing, i.e., editing multiple genes simultaneously using different sgRNAs8.
[4] See here for a fantastic review of the therapeutic applications of CRISPR and gene editing systems